Archive Site Provided for Historical Purposes

Sponsored by the U.S. Department of Energy Human Genome Program
Human Genome News, April-June 1996; 7(6)
Santa Fe 96
Last year's triumphs in sequencing entire microbial genomes [HGN 7(1), 5 (May-June 1995)] left little doubt that the era of large-scale sequencing had begun. The latest whole-genome sequencing feat was presented at the workshop, this time for the heat-loving, methane-producing M. jannaschii.
These remarkable accomplishments represent an important step toward developing and optimizing the technologies and strategies needed to fully sequence the 3 billion bases of human DNA. Optimism runs high that the first human genome reference sequence can be obtained on time, even without revolutionary technical advances. However, significant improvements still are needed for increasing the accuracy and efficiency and reducing the cost of conventional gel-electrophoretic methods.
Current large-scale genome sequencing focuses on using gel-based instruments with random (shotgun) or directed strategies, often combining elements of both approaches. Pilot projects for production sequencing of human DNA concentrate on identifying and analyzing areas of known biological importance. (See articles on LANL, LLNL, and LBNL in "Progress at the DOE Labs"; on the comparative analysis of human and mouse in "Leaping Across Genomes"; and on the NIH program to establish six large-scale pilot human genome sequencing centers, in NCHGR Initiates Sequencing Pilot Projects.)
Support for the whole-genome and other microbial sequencing efforts described below comes from DOE's new Microbial Genome Program, which aims to sequence the genomes of microbes with potential industrial, environmental, and economic importance.
In addition to microbial production-sequencing reports, speakers presented progress on technology improvements with current gel-based systems; improved gel technologies, especially capillary electrophoresis (CE); and alternative, potentially high payoff technologies such as mass spectrometry (MS). Many of these DNA-analysis technologies will feed into the rapidly expanding biotechnology industry, where they will have broader applications in clinical diagnostics, environmental testing, industrial process monitoring, forensics, and agriculture.
Highlights of presentations follow.
Microbial Genome Sequencing
M. jannaschii
Carol Bult [The Institute for Genomic Research (TIGR)] described the whole-genome shotgun-sequencing strategy used to obtain the complete genomic sequence of M. jannaschii, an organism first isolated from a deep-sea hydrothermal vent in 1982. This genome is the first to be completed from the Archaea domain of life, a group of unique microbes that are genetically distinct from both bacteria and eukaryotes. The Archaea, which include methanogens, thermoacidophiles, and extreme halophiles, may represent some of the earliest forms of living cells.
Bult identified several aspects critical to the group's success. These factors include the availability of a random genomic 2.5-kb-insert plasmid library from Gary Olsen (University of Illinois)and a representative 20-kb-insert lambda library for building a genome scaffold; high-quality sequence data from both ends of the plasmid and lambda clones (using ABI 373 and 377 automated sequencers with fluorescent technologies); and a robust sequence fragment assembly engine (TIGR Assembler, discussed below by G. Sutton). Sequence coverage was obtained for the entire genome. Bult emphasized the importance of tightly integrating data production with tools for managing and analyzing data. Sequence annotation is now complete, and all data and clones will be available by early summer (see TIGR WWW page, http://www.tigr.org/).
Pyrococcus furiosus
Robert Weiss (University of Utah) described a project to sequence the 2-Mb genome of P. furiosus, another of the hyperthermophilic Archaea. Investigators are using a multiplexed, transposon-based directed approach with an end-sequencing strategy. In both the mapping and sequencing phases, automated devices detect enzyme-linked fluorescence from DNA hybrids on nylon membranes.
Summarizing mapping progress, Weiss reported that 1.4 Mb (two-thirds of the genome) had been processed; 2500 mapped clones, representing minimal-set coverage of 0.76 Mb, have been sequenced. Mapping transposons, he noted, is much simpler than performing the base-calling, assembly, and editing steps required by other sequencing approaches, and the integrity of the transposon map encourages scaleup. The group is now in the end-sequencing phase and will piece together the genome by aligning end sequence with nucleated transposon maps and walking their way to continuity. They have about 500 kb of consensus sequence.
Borrelia burgdorferi
John Dunn [Brookhaven National Laboratory (BNL)] and colleagues are sequencing the 935-kb genome of B. burgdorferi, the spirochete that causes Lyme disease, to develop methods for DNA sequencing by primer walking with a hexamer library. The goal is to eliminate up-front mapping and enable a tenfold reduction in the template preparations required for a typical shotgun project. The approach involves using end sequencing and either hexamer strings or ligated hexamers (18-mers) to walk down the molecule. As a test of the method, a 35-kb Borrelia fesmid clone (based on the Fos vector) was sequenced by this approach. Dunn believes the group can achieve fourfold redundancy, with both strands completely sequenced. The next stage in the project, he said, is to bring online a high-volume CE system to allow faster sequencing ( http://www.biology.bnl.gov/cellbio/dunn.html).
Sequencing Technologies
Researchers at the workshop reported on efforts to optimize current approaches in scaling up for multi- megabase sequencing. Goals are to minimize or eliminate some of the bottlenecks at each step in the sequencing process: template isolation, sequencing reactions, fragment separation and detection, and data collection and analysis.
Emphasis is on developing fully automated, integrated, modular systems that can process a sample from template isolation to data analysis with little or no human intervention.
Front-End Automation: The "Sequatron"
The "Sequatron" described by Trevor Hawkins (Whitehead-MIT) uses available components to automate and integrate the tasks of DNA isolation, setup of sequencing reactions,thermal cycling, and sample purification and concentration for separation on gels. The major component is an articulated CRS 255A robotic arm. Solid-phase reversible immobilization is used to isolate and manipulate the DNA on magnetic particles throughout the process.
Current throughput is 80 microtiter plates of samples (about 8000) from M13 phage supernatants or crude PCR products to sequence-ready samples every 24 hours. New enzymes, energy-transfer primers, and higher-density microtiter plates may increase throughput to 25,000 samples.
Speeding Up DNA Separation and Detection: Capillary Gel Systems
Researchers reported surprising and exciting results over the past year in developing newer CE methods for dramatically faster and higher-resolution DNA fragment separation. Advantages of CE include longer read lengths; improved heat transfer in the long, thin, polymer-filled capillaries vs that in standard slab gels; automatability; and online fragment detection.
Edward Yeung (Iowa State University)described a CE system based on novel separation, detection, and imaging techniques for real-time monitoring that he believes will soon enable sequencing of 40 Mb of DNA in a single day. A first-generation system has been constructed that uses new fluid-gel matrices in 100 capillaries that are read simultaneously in real time, compared with the much slower consecutive-reading technology available with current instruments. Most processes are automated. The team is scaling up the technology to allow parallel sequencing in up to 1000 capillaries.
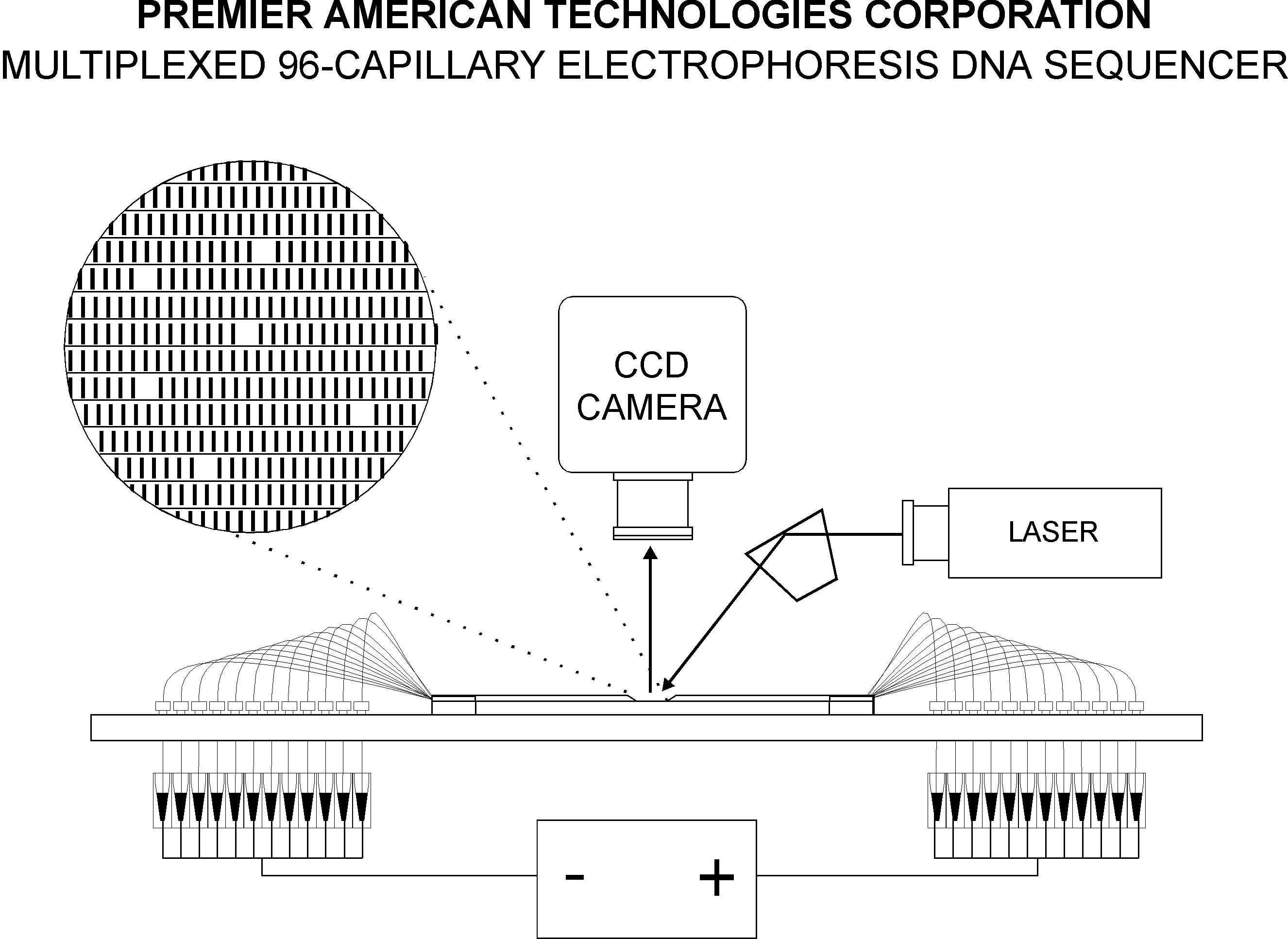
click to enlarge
Multiplexed CE DNA sequencer developed by Premier American Technologies Corp. (Bellefonte, PA). The sequencer uses 96 individual capillaries and a single-laser-CCD detection system. The ESY9600 is designed for routine, simultaneous analysis of 96 DNA samples (400+ bases each).
The new system, the ESY9600 Multiplexed Capillary Electrophoresis DNA Sequencer, is scheduled for release this year by Premier American Technologies Corporation, which licensed the technology from DOE's Ames Laboratory at Iowa State. The system uses cartridges of 96 fused-silica capillaries in the electrophoresis and a PC with software for automation control and data processing. Fragments are detected using an argon laser and a CCD camera. The new system offers a 100-fold gain in speed and 24-fold gain in throughput over conventional automated sequencers.
Norman Dovichi (University of Alberta, Edmonton) achieved reads of 1000 bases in 135 min. by decreasing the electric field to 100 to 150 V/cm, increasing temperature to 70°C, and using a noncrosslinked polyacrylamide matrix (3% concentration) in the capillaries. His presentation focused on high-sensitivity fluorescence-detection cuvettes made with pyrex microscope slides into which are etched a series of fingers that align precisely with the capillaries. The 16-capillary machine is now online, and the group has demonstrated success with a two-dimensional array design (4-by-5 and 8-by-12 rows of capillaries), for which they recently filed a patent. They are now working on a 24-by-24 array (576 capillaries) that has the potential to generate 500 bases per run or up to 300 Mb of raw sequence in about 2 hours. With 4 runs a day, this could yield 1 Mb of sequence per day per instrument.
Barry Karger (Northeastern University) updated progress toward production of a robust CE system using replaceable polymer matrices. Factors to optimize include type and concentration of polymer matrix, column conditions, dye chemistries, and software. Karger's group found that low (2%) concentrations of very high molecular-weight replaceable noncrosslinked polymer, elevated temperatures, and signal-processing software allow fast sequencing and long reads (1000 bases in 80 min. with 97% accuracy). A multiple capillary-array system with CCD camera currently is being tested with a goal of 1 kb per column every 1.5 hours.
Richard Mathies [University of California, Berkeley (UCB)] discussed his collaboration with Alexander Glazer (UCB) and others to develop new DNA detection methods and CE chips for ultrahigh-speed DNA analysis. Jingyue Ju, former Human Genome Postdoctoral fellow now at Incyte Pharmaceuticals (Palo Alto, California), reported on the development of high-sensitivity energy-transfer dye-labeled primers for DNA sequencing and PCR analysis. Mathies and Glazer are using four sets of these dyes to perform four-color confocal sequencing of mitochondrial DNA in CE. They are also developing a miniaturized integrated DNA analysis system (MIDAS), combining their CE arrays and Allen Northrup's (LLNL) fast PCR systems. Mathies envisions applying this technique to samples generated by cycle sequencing to produce and analyze 15 kb per chip per hour.
Mark Quesada (BNL) described a prototype 8-capillary DNA sequencer that uses replaceable linear polyacrylamide matrices and fiber optics for illumination and detection. Resolution limits are about 400 to 450 bases per hour. The team hopes to have a 21-capillary system this summer to use in the directed sequencing project aimed at the genome of the microorganism that causes Lyme disease (see J. Dunn above).
Innovative Gel-Less Technologies: Mass Spectrometry
Replacing the gel-separation step with MS promises resolution, accuracy, and speed surpassing those of electrophoretic methods. MS uses the difference in mass-to-charge ratios of ionized atoms or molecules to separate them. Problems are encountered in ionizing large DNA fragments with minimum sample degeneration.
Richard Smith (Pacific Northwest National Laboratory) and Lloyd Smith (University of Wisconsin) talked about the challenges and potential capabilities of DNA sequencing using MS with electrospray (ES) ionization and matrix-assisted laser desorption ionization (MALDI).
The gentler, solution-based ES ionization produces ions with a distribution of net charge states that spreads the molecule signal among several mass-spectrometric peaks, complicating interpretation of the resulting mass spectrum. This problem is compounded by salts associating with the molecule during the sample-preparation process. Richard Smith reported striking reduction of salt content by incorporating an online microdialysis unit before the ionizing step. Either single- or double-stranded PCR products of >100 bp could be analyzed and accurate mass measurements obtained without any detectable fragmentation that would make sequencing much more difficult. The researchers also demonstrated a method for dynamic range expansion using Fourier transform ion cyclotron resonance MS that significantly extends the useful read length for sequencing.
Lloyd Smith's laboratory is exploring reasons for DNA fragmentation during MALDI from a solid matrix. Fragmentation is dependent on sequence as well as matrix, and chemical modification may limit the breakage. Smith's group identified a base-protonation mechanism they believe is involved in strand breakage, and the team is now developing a set of modified nucleotides that could offer protection.
Sequence Finishing and Analysis
Sequence Finishing
A key component in high-throughput sequencing systems will be the integration of improved software tools for collecting and finishing the sequence. Steps in this process include automated reading of sequencing gels (base calling), assembly of contiguous sequence from separate DNA fragments (sequence assembly), and editing to resolve ambiguities.
Many assembly algorithms are available, but they cannot handle data sets with large repeat regions. Other assembly challenges include large numbers of pairwise comparisons to determine overlaps in large data sets, sequence-assembly uncertainties due to clone chimerism, and sequencing errors. Granger Sutton (TIGR) described TIGR Assembler, which was used to assemble the shotgun-sequenced genomes of H. influenzae, M. genitalium, and M. jannaschii. It is also being used to assemble shotgun-sequenced BAC clones. Source code and executables for TIGR Assembler are available to nonprofit researchers(grange@tigr.org).
Phil Green (University of Washington, Seattle) discussed two software tools that improve accuracy and minimize human involvement. Phred improves base calls substantially and assesses the quality of processed ABI 373A and 377 trace data. Phrap is a sequence-assembly program that uses information from Phred and from read comparisons to delineate promising base calls; this helps identify repeats and allows use of the full reads in assembly. Phrap also identifies data anomalies such as chimeras and vector DNA and will soon incorporate mapping information. Green's group is nearing the point at which unedited, automatically assembled sequences will give an error rate of 1 per 10 kb on typical cosmid data sets. Phred and Phrap are now being beta tested.
Noting that sequence accuracy improves exponentially with incremental project cost, David States (Washington University, St. Louis) argued for setting accuracy criteria when large-scale projects are implemented. The ultimate usefulness of a sequence to the wider biochemical and clinical communities is an important consideration, he said, noting that an error rate of 1 in 104 would be achievable and tolerable. States' team is pursuing a mathematical, model-based approach to the entire sequencing process and has completed a nonproprietary data path from gel images through finished sequence as prototype code. In a panel discussion, States noted that the group's lane-tracking software is available from their ftp site and that trace extraction and further code, still in the developmental stage, is accessible to collaborators.
Sequence Analysis
Computers sift stretches of DNA sequence for patterns that identify such biologically important features as protein-coding regions, regulatory areas, and RNA splice sites. Other computer tools are used to compare a new sequence (i.e., a gene) against all other entries in a database and retrieve any homologous sequences that have already been entered.
The popular GRAIL and GenQuest servers localize genes and other biologically important features in sequence and search other databases for homologous DNA and protein sequences and structural motifs. Over 17 million bases of sequence are processed each month in 13,000 sessions. GRAIL, a comprehensive system that uses artificial intelligence and machine learning to recognize many different signals, allows the system to incorporate more complex relationships between the data than might be anticipated a priori. GenQuest takes information generated by GRAIL and compares it with data in protein, DNA, and motif databases.
Ed Uberbacher and Richard Mural (both at Oak Ridge National Laboratory) presented the latest version (1.3) of GRAIL. This version features improved sensitivity and splice-site accuracy, better performance in AT-rich regions, new analysis systems for four model organisms, frameshift detection, batch processing, and a wider variety of ways to access GRAIL automatically or interactively. GRAIL 1.3 also builds annotation reports; processing a cosmid or an even larger sequence can be done in under an hour. Responding to requests for a more automated approach for GRAIL analysis, the group will provide UNIX socket access to all functions of the new version (http://compbio.ornl.gov/Grail-1.3/).
Randy Smith [Baylor College of Medicine (BCM)] described efforts to improve user access to the wide variety of database-search tools available on WWW. The BCM Search Launcher features a single point of entry for related searches, the addition of hypertext links to results returned by remote servers, and a batch client.
Smith outlined other activities of the BCM group. FASTA-SWAP, a new pattern-search tool for databases, improves sensitivity and specificity to help detect related sequences. BEAUTY, an enhanced version of the BLAST database-search program, improves access to information about the functions of matched sequences and incorporates additional hypertext links. The graphical displays allow correlation of hit positions with annotated domain positions.
Future plans include developing a post-processor version of BEAUTY and providing access to information from and direct links to other databases, including organism-specific databases. The BCM group is also furnishing external analysis services to the Genome Sequence Data Base sequence annotator. Human Genome Postdoctoral fellow Mark Graves (BCM) reported on a simple database-management system for biologists to use in designing their own laboratory databases (mgraves@bcm.tmc.edu).
Gary Stormo (University of Colorado) described an approach for predicting coding regions in genomic DNA; it uses multiple types of evidence, combines them into a single scoring function, and returns both optimal and ranked suboptimal solutions. The approach is robust to substitution errors but sensitive to frameshift errors. Stormo's group is now exploring methods for predicting other classes of sequence regions, especially promoters.
Return to the Table of Contents
The electronic form of the newsletter may be cited in the following style:
Human Genome Program, U.S. Department of Energy, Human Genome News (v7n6).
The Human Genome Project (HGP) was an international 13-year effort, 1990 to 2003. Primary goals were to discover the complete set of human genes and make them accessible for further biological study, and determine the complete sequence of DNA bases in the human genome. See Timeline for more HGP history.

Published from 1989 until 2002, this newsletter facilitated HGP communication, helped prevent duplication of research effort, and informed persons interested in genome research.
