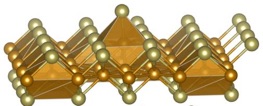
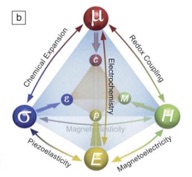
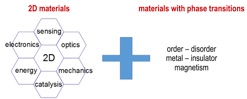
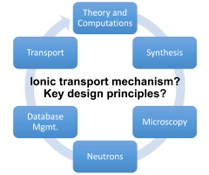
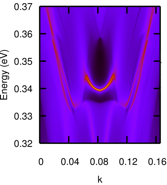
Research and Development Staff Scientist
Center for Nanophase Materials Sciences
Oak Ridge National Laboratory
Tel: 1-865-574-1999
Fax:1-865-574-1999
Email: ganeshp AT ornl.gov


Research and Development Staff Scientist
Center for Nanophase Materials Sciences
Oak Ridge National Laboratory
Tel: 1-865-574-1999
Fax:1-865-574-1999
Email: ganeshp AT ornl.gov

© Copyright, P. Ganesh, ORNL, Wednesday, August 28, 2019