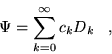The basis for CI methods is the simple
observation that an exact many-body wavefunction, ![]() , may be
written as a linear combination of Slater determinants,
, may be
written as a linear combination of Slater determinants, ![]() ,
,
The Hartree-Fock ``reference determinant'' ![]() is by definition the best
single-determinant approximation to the exact wavefunction
is by definition the best
single-determinant approximation to the exact wavefunction ![]() . In
most electronic systems, the Hartree-Fock energy accounts for the
majority of the exact total energy, and the missing correlation energy
is small. If the coefficients
. In
most electronic systems, the Hartree-Fock energy accounts for the
majority of the exact total energy, and the missing correlation energy
is small. If the coefficients ![]() are normalised then typically
are normalised then typically
![]() and all remaining
and all remaining ![]() are very small. A very large
number of configurations is required to yield energies and
wavefunctions approaching the exact many-body wavefunction. In
practice the expansion must be limited on physical grounds, as the total
number of determinants is
are very small. A very large
number of configurations is required to yield energies and
wavefunctions approaching the exact many-body wavefunction. In
practice the expansion must be limited on physical grounds, as the total
number of determinants is
| (2.11) |
The scientific problem in adapting the CI method into a practical one is to obtain the best wavefunction, and hence lowest CI energy, with the shortest expansion length. A typical approach would be to truncate the expansion after only double or quadruple excitations from the reference determinant, where an excitation consists of replacing a ground state occupied orbital by an unoccupied one. These levels of truncation are the CI singles-doubles (CISD) and CI singles-doubles-triples-quadruples (CISDTQ) methods. A formidable number of terms are still left in the expansion. Accurate applications of the methods are consequently limited due to their computational cost.
When performed within a finite reference space, an additional problem with the method becomes apparent: the methods lack ``size-extensivity'' and do not perform equally well in systems of differing size. As the size of system increases, the proportion of the electronic correlation energy contained within a fixed reference space (such as all single and double excitations) decreases. The lack of size-extensivity results in a non-cancellation of errors when systems of different sizes are compared, resulting in difficulties when interaction or bonding energies are required.
Despite these limitations, CI represents a controlled (and variational) improvement to the ground-state wavefunction, and may therefore be used in the determinantal parts of trial wavefunctions in QMC. Details of this approach are given in chapter 3.
The coupled-cluster (CC) method[7,3] is
one of the most important practical advances over the CI
method. Although non-variational, it resolves the problem of size
extensivity, and is often very accurate, but more expensive than
(limited) CI. The CC method assumes an exponential ansatz for the
wavefunction
| (2.12) |
| (2.13) |
| (2.14) |
A ``coupled-cluster doubles'' wavefunction is written as
| (2.15) |
| (2.16) |
The key limitation of the CC methods are their rapid increase in computational cost with system size. This presently limits the methods to small molecular systems.
Almost all post Hartree-Fock methods share the combined limitations of a poor scaling with system size and a strong basis set dependence. In practice, post Hartree-Fock methods typically scale with the fourth or higher power of the number of basis states included in the calculation, limiting their application to small systems. The basis states depend on the underlying basis set used in their numeric expansion, equation 2.9, and it is commonly found that use of improved methods requires an improved basis set, further increasing the cost of calculation. CI and CC-based methods effectively transform the electron-correlation problem into a basis set problem, where the basis set is the set of molecular orbitals derived from a Hartree-Fock (or similar) calculation.