


Next: 3.5 Summary
Up: 3. Quantum Monte Carlo
Previous: 3.3 Variational Monte Carlo
Contents
Subsections
Diffusion Monte Carlo (DMC) is a projector or Green's function based
method for solving for the ground state of the many-body
Schrödinger equation. In principle the DMC method is exact,
although in practice, several well-controlled approximations must be
introduced for calculations to remain tractable.
The DMC method is based on rewriting the Schrödinger
equation in imaginary time, 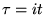 . The imaginary time
Schrödinger equation is:
. The imaginary time
Schrödinger equation is:
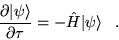 |
(3.25) |
The state 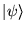 is expanded in eigenstates of the
Hamiltonian
is expanded in eigenstates of the
Hamiltonian
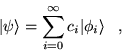 |
(3.26) |
where
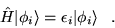 |
(3.27) |
A formal solution of the imaginary time
Schrödinger equation is
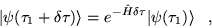 |
(3.28) |
where the state evolves from imaginary time 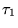 to
a later time
to
a later time
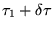 . If the initial state
. If the initial state
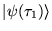 is expanded in energy ordered eigenstates,
following equation 3.26, then
is expanded in energy ordered eigenstates,
following equation 3.26, then
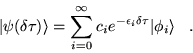 |
(3.29) |
Hence any initial state, , that is not
orthogonal to the ground state,
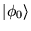 , will evolve to the
ground state in the long time limit:
, will evolve to the
ground state in the long time limit:
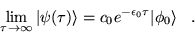 |
(3.30) |
This derivation shares many formal similarities with that
given for the variational principle in
section 3.3.1. However in the DMC method the imaginary time
evolution results in excited states decaying exponentially fast,
whereas in the VMC method any excited state contributions remain and
contribute to the VMC energy.
The DMC method is a realisation of the above derivation in position space.
equation 3.30 becomes
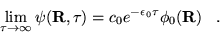 |
(3.31) |
By introducing a constant offset to the energy,
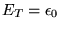 ,
the long-time limit of equation 3.31 can be kept finite.
If the Hamiltonian is separated into the kinetic energy and potential
terms, the imaginary time Schrödinger equation,
equation 3.25, takes on a form similar to a diffusion equation:
,
the long-time limit of equation 3.31 can be kept finite.
If the Hamiltonian is separated into the kinetic energy and potential
terms, the imaginary time Schrödinger equation,
equation 3.25, takes on a form similar to a diffusion equation:
![\begin{displaymath}
-\frac{\partial \psi ({\bf R}, \tau )}{\partial\tau}=
\left[...
... R},\tau)\right]
+(V({\bf R})-E_{T})\psi({\bf R},\tau) \;\;\;.
\end{displaymath}](pkthimg227.gif) |
(3.32) |
This equation is a diffusion equation where 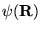 may be
interpreted as the density of diffusing particles (or ``walkers''),
and the term
may be
interpreted as the density of diffusing particles (or ``walkers''),
and the term
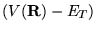 is a rate term describing a
potential-dependent increase or decrease in the particle density. This
is shown schematically in one-dimension for a simple potential in
figure 3.1.
is a rate term describing a
potential-dependent increase or decrease in the particle density. This
is shown schematically in one-dimension for a simple potential in
figure 3.1.
Figure 3.1:
The evolution of
walkers in the DMC method. A single particle in confined in a simple
potential well, 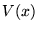 . An initially even distribution of walkers
(circles) is propagated in imaginary time,
. An initially even distribution of walkers
(circles) is propagated in imaginary time, 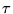 . The distribution
gradually evolves by a process of diffusion and branching to a
distribution representative of the ground state wavefunction,
. The distribution
gradually evolves by a process of diffusion and branching to a
distribution representative of the ground state wavefunction,
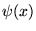 . Note that where the potential energy is low, walkers tend
to branch giving a higher density of walkers, and where the potential
energy is high, walkers tend to be removed.
. Note that where the potential energy is low, walkers tend
to branch giving a higher density of walkers, and where the potential
energy is high, walkers tend to be removed.
|
|
The above equation may be transformed into a form suitable for Monte
Carlo methods, [23,27] but this
leads to a very inefficient algorithm. The potential 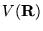 is
unbounded in coulombic systems and hence the rate term,
is
unbounded in coulombic systems and hence the rate term,
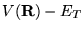 , can diverge. Large fluctuations in the particle density
then result and give impractically large statistical errors. These
fluctuations may be substantially reduced by the incorporation of
importance sampling in the algorithm.
, can diverge. Large fluctuations in the particle density
then result and give impractically large statistical errors. These
fluctuations may be substantially reduced by the incorporation of
importance sampling in the algorithm.
The above method also lacks the important constraint that for
electronic systems the solution should be antisymmetric with respect to the exchange of any two electrons, i.e. is fermionic. This constraint
may be imposed by assuming a form for the nodal surface of the
fermionic wavefunction, but at the expense of obtaining only a
variational solution to the exact ground state energy. This method of
constraint, the fixed-node approximation, and other methods of
enforcing fermionic anti-symmetry are discussed in
section 3.4.4.
Importance sampling is essential for DMC methods, if the
simulation is to be efficient. [27] A trial or guiding wavefunction,
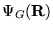 , which closely approximates the ground state
wavefunction is introduced. The derivation of Reynolds
et. al.[28,23] is
followed in the remainder of this section.
, which closely approximates the ground state
wavefunction is introduced. The derivation of Reynolds
et. al.[28,23] is
followed in the remainder of this section.
A new distribution is defined
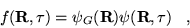 |
(3.33) |
which is also a solution of the Schrödinger equation when
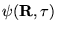 is a solution. The equation for non-importance
sampled DMC, equation 3.32, is consequently modified:
is a solution. The equation for non-importance
sampled DMC, equation 3.32, is consequently modified:
![\begin{displaymath}
-\frac{\partial f({\bf R},\tau)}{\partial\tau}=
\left[\sum_{...
...R},\tau)\right]
+(E_{L}({\bf R})-E_{T})f({\bf R},\tau) \;\;\;.
\end{displaymath}](pkthimg239.gif) |
(3.34) |
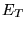 is a ``trial energy'' introduced to maintain normalisation of the projected solution at large .
The term
is a ``trial energy'' introduced to maintain normalisation of the projected solution at large .
The term
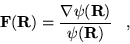 |
(3.35) |
is commonly referred to as the ``quantum
force''[29]. The local energy,
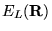 ,
equation 3.17 is computed with respect to the guiding wavefunction
,
equation 3.17 is computed with respect to the guiding wavefunction
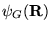 .
.
The right hand side of the importance sampled DMC equation (equation
3.34) consists, from left to right, of diffusion, drift
and rate terms. The problematic potential dependent rate term of the
non-importance sampled method is replaced by a term dependent on the
difference between the local energy of the guiding wavefunction and
the trial energy. The trial energy is initially chosen to be the VMC
energy of the guiding wavefunction, and is updated as the simulation
progresses. Use of an optimised guiding function minimises the
difference between the local and trial energies, and hence minimises
fluctuations in the distribution 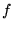 . A wavefunction optimised using
VMC is ideal for this purpose, and in practice VMC provides the best
method for obtaining wavefunctions that accurately approximate ground
state wavefunctions locally. The guiding wavefunction may be also
constructed to minimise the number of divergences in
,
unlike the non-importance sampled method where divergences in the
coulomb interactions are always present.
. A wavefunction optimised using
VMC is ideal for this purpose, and in practice VMC provides the best
method for obtaining wavefunctions that accurately approximate ground
state wavefunctions locally. The guiding wavefunction may be also
constructed to minimise the number of divergences in
,
unlike the non-importance sampled method where divergences in the
coulomb interactions are always present.
3.4.3 Transformation to integral form
To be of use, the importance sampled DMC equation equation
3.34 must be transformed into a form suitable for Monte
Carlo integration. The transformation is more complex than for VMC,
which simply required the insertion of the factor
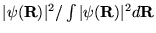 into the conventional formulas
for quantum mechanical expectation values.
into the conventional formulas
for quantum mechanical expectation values.
A Green's function,
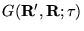 , that is a
solution of equation 3.34 is desired, i.e, a spatial
representation of the imaginary time propagator,
, that is a
solution of equation 3.34 is desired, i.e, a spatial
representation of the imaginary time propagator,
![$\exp[-\tau(\hat{H}-E_{T})]$](pkthimg245.gif) :
:
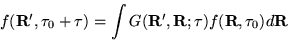 |
(3.36) |
The Green's function may be approximated
to second order in by factorising the propagator into branching
and diffusion parts: [28,23]
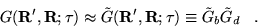 |
(3.37) |
The branching part of the Green's function, 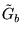 , is a
function of the ``surplus local energy'':
, is a
function of the ``surplus local energy'':
![\begin{displaymath}
\tilde{G}_{b}({\bf R}^\prime,{\bf R};\tau) =
\exp[(-\frac{1}{2}(E_{L}({\bf R}^\prime)+E_{L}({\bf R}))-E_{T})\tau] \;\;\;.
\end{displaymath}](pkthimg249.gif) |
(3.38) |
The diffusion part is obtained by assuming that the quantum force,
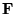 , is constant over the move
, is constant over the move
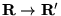 :
:
![\begin{displaymath}
\tilde{G}_{d}({\bf R}^\prime,{\bf R};\tau) =
(2\pi \tau)^{-3...
...f R}^\prime-\tau {\bf F}({\bf R}^\prime)/2)^{2}/{2\tau}] \;\;.
\end{displaymath}](pkthimg252.gif) |
(3.39) |
Although accurate to second order in , this approximation is
significant in all-electron calculations, where varies
rapidly in core regions. Additionally, this component of the
short-time approximation Green's function, 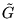 , is not
Hermitian. ``Detailed balance'' is imposed with the conventional
Metropolis probability
, is not
Hermitian. ``Detailed balance'' is imposed with the conventional
Metropolis probability
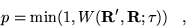 |
(3.40) |
where
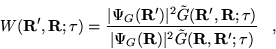 |
(3.41) |
3.4.4 The fixed-node approximation
The DMC method discussed so far lacks a constraint crucial for the
simulation of fermionic systems. In general, the ground state
wavefunction of a fermionic system has nodes and consequently regions
of positive and negative sign. These regions are essential for a
wavefunction to be anti-symmetric with respect to the interchange of
any two fermions. The methods discussed so far rely on a probability
or particle distribution which by definition is positive definite. If
the constraint of antisymmetry is not imposed, the application of the
DMC method results in a bosonic solution of lower energy than the true
fermionic ground state energy being obtained. The most successful
attempt to address to this ``nodal problem'' is the fixed-node
approximation. It is used in all of the DMC calculations presented in
this thesis.
The fixed-node approximation, due to Anderson, [30]
was the first solution of the nodal problem. The approximation consists
of imposing the nodes of a reference wavefunction. This approximation
is variational: the fixed-node DMC energy is an upper bound to the
exact ground state energy.[31] In Anderson's original
implementation, walkers attempting to cross nodes were deleted. This
has been shown to introduce a bias that is linear in the time
step, [29] and all current implementations reject
moves that cross nodes. A bias of second order in the time step
results, which is the same order as the short-time approximation to
the Green's function.
The accuracy of the fixed-node approximation is wholly dependent upon
the nodes of the reference wavefunction. In practice the nodes of an
HF or DFT based wavefunction are found to be very accurate, giving
energies well below those of the best VMC wavefunctions. This is fortunate
as currently there are no general methods for systematically improving
the DMC energy that scale polynomially with the size of the system.
Two methods for systematically improving the fixed-node energy exist:
- The release-node method[31] is a simple refinement
of the fixed-node method. An antisymmetric wavefunction is obtained by
averaging the difference between populations of negative and positive
walkers, initially ``equilibrated'' within the fixed-node
approximation. Moves that cross node are then permitted and the
asymptotic release-node energy obtained. This method is impractical
for more than a few electrons as the populations and hence statistical
errors grow exponentially. The fixed-nodes must lie close to the exact
nodes and the system must rapidly relax to the exact fermionic
solution for the method to be practical, even in small systems. The
most successful application of this technique is for the electron
gas. [13]
- The nodes of CI-based wavefunctions can be expected to more
accurately represent the exact nodes than HF-based wavefunctions, as
correlation effects are partly included and a CI wavefunction is a
better approximation to the exact wavefunction. Therefore a
systematic, but inefficient, method of improving the nodes of the
reference wavefunction is to perform large CI calculations and use the
resulting wavefunction (with many determinants) in DMC. This method is
impractical for systems of moderate size for the same reasons that the
CI approach is in general impractical (see section 2.4.1). For
small systems this approach is practical, but has not been extensively
employed in the literature.
A third method of improving the nodes
exists. Calculations by Kwon[32,33] have shown
that small improvements to DMC energies may be obtained for the 2D and
3D electron gases, at high densities, by replacing the positions of the
electrons in the wavefunctions by ``quasiparticle coordinates'' [34]
parameterised in terms of the positions of (in principle) all the
electrons:
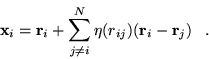 |
(3.42) |
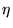 is a correlation or backflow[34]
function containing variable parameters. Presently it is unclear
whether the use of quasiparticle coordinates will be of use in real
electronic systems. These methods carry considerable overhead, as the
full wavefunction must be calculated when a single electron is
moved. Although these methods may prove useful in small systems, the
use of quasiparticle coordinates in large systems is likely to
restricted, as the cost of moving all of the electrons is now
is a correlation or backflow[34]
function containing variable parameters. Presently it is unclear
whether the use of quasiparticle coordinates will be of use in real
electronic systems. These methods carry considerable overhead, as the
full wavefunction must be calculated when a single electron is
moved. Although these methods may prove useful in small systems, the
use of quasiparticle coordinates in large systems is likely to
restricted, as the cost of moving all of the electrons is now 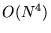 due to the additional cost of wavefunction recalculation (see chapter
4).
due to the additional cost of wavefunction recalculation (see chapter
4).
Additionally, these methods are perturbative in the sense that they
modify an underlying reference nodal set. Although the use of
quasiparticle coordinates may result in small improvements in real
electronic systems, where good quality nodal sets are already
available, their use cannot be considered a solution to the
nodal problem. They are however, one of the few ideas present in the
literature that scales reasonably well with system size.
3.4.6 The DMC algorithm
The DMC algorithm used for most of the calculations in this thesis is
given below. It is similar to those of
Refs. [28,29,23].
As with VMC there are distinct equilibration and accumulation phases,
but both are almost identical as energies are required at every move
in the calculation in the evaluation of the configuration branching
factor.
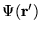 is used to indicate the wavefunction with a
single electron moved to point
is used to indicate the wavefunction with a
single electron moved to point
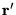 and all other
electrons unmoved:
and all other
electrons unmoved:
- Initialise an ensemble of
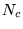 configurations, which should
be uncorrelated and distributed according to the probability density
of the guiding function
configurations, which should
be uncorrelated and distributed according to the probability density
of the guiding function 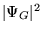 . Initialise the trial energy
to the average VMC energy of the ensemble.
. Initialise the trial energy
to the average VMC energy of the ensemble.
- For every configuration
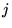 :
:
- For every electron
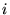 in the current configuration:
in the current configuration:
- Propose a move according to
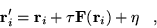 |
(3.43) |
where is the ``quantum force'' of equation 3.35, and 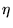 is a Gaussian random vector of mean zero and variance per
Cartesian component.
is a Gaussian random vector of mean zero and variance per
Cartesian component.
- Apply the fixed-node approximation, checking that
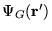 is of the same sign as
is of the same sign as
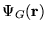 . If a node has
been crossed, reject the move of electron and consider the next
electron.
. If a node has
been crossed, reject the move of electron and consider the next
electron.
- Compute the weight for the move
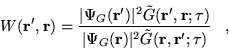 |
(3.44) |
where is the short-time approximation to the Green's
function (section 3.4.3).
- Accept the move according to the Metropolis probability
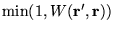
- Compute the branching factor for the moved configuration :
![\begin{displaymath}
P_{B} = \exp\left(-\tau
(\frac{1}{2}\left[E_{L}({\bf R}^\prime)+E_{L}({\bf R})\right]-E_{T})\right)
\end{displaymath}](pkthimg268.gif) |
(3.45) |
- Accumulate the energy and any observables weighted by the
branching factor
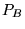
- Make
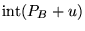 copies of the configuration, where
copies of the configuration, where
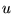 is a uniformly distribution random number of
is a uniformly distribution random number of ![$[0,1]$](pkthimg271.gif) .
.
- Repeat the configuration moves for a number of steps (a block), typically
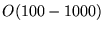 .
.
- Update the trial energy , to bring it closer to that of
the current ensemble, by using the average energy for the last block.
- Renormalise the number of walkers in the ensemble to the target
number
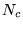 by randomly creating or deleting walkers.
by randomly creating or deleting walkers.
- Repeat until sufficient data is obtained.
During equilibration, a slight variant of the above algorithm is used, where
the trial energy is updated more frequently. While
equilibrating, there is tendency for the number of walkers to increase
exponentially as the excited eigenstates in the trial wavefunction are
propagated out. The trial energy is replaced by a reference energy
updated after every move of all configurations:
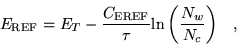 |
(3.46) |
where
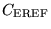 is a system dependent parameter set to
control the fluctuations in the current number of configurations
is a system dependent parameter set to
control the fluctuations in the current number of configurations
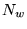 compared with the target number of configurations .
compared with the target number of configurations .
Several improvements to the above algorithm have been
proposed. [29] These modify the Green's function where
the short time approximation is expected to be least accurate
(adjacent to atomic cores and near nodes), and also take account of
the modified timestep due to the rejection of some of the single
electron moves. These improvements give a significant reduction in the
timestep error in all electron calculations. In pseudopotential
calculations the improvements are much smaller. Small
efficiency improvements are possible by assigning weights to each
walker, and only branching or deleting walkers when the weights extend
beyond a defined range. Provided sufficiently small timesteps
are used with sufficiently accurate wavefunctions, these different
algorithms give the same ground state energy.
Next: 3.5 Summary
Up: 3. Quantum Monte Carlo
Previous: 3.3 Variational Monte Carlo
Contents
© Paul Kent
![\includegraphics [width=10cm]{Figures/branch.eps}](pkthimg230.gif)