Archive Site Provided for Historical Purposes

Sponsored by the U.S. Department of Energy Human Genome Program
Human Genome News, September-December 1995; 7(3-4):4
This article describes Human Genome Project accomplishments and progress toward short and long-term goals. Topics include genetic and physical mapping of the human genome; DNA sequencing; gene identification; analysis of model organism genomes; informatics; and explorations of ethical, legal, and social implications (ELSI) arising from genome research.
GENETIC MAPPING
Genetic linkage maps are critical for mapping genes underlying identifiable phenotypes including diseases. In late 1994 the first major initial genome project goal, a 2- to 5-cM human genetic map, was reached when an international group of investigators published a comprehensive map comprising 5840 loci covering 4000 cM. Of those markers, 970 are ordered with high confidence (odds of >1000:1) and provide a framework map. The comprehensive map can be said to represent an average marker density of 0.7 cM, with the more highly reliable framework map subset having a resolution of about 4 cM.
Progress toward this goal was extremely rapid; the map was, in fact, completed a year ahead of schedule. This accomplishment resulted, in part, from the use of a new type of genetic marker known variously as a microsatellite repeat, STRP, or SSLP. Advantages of microsatellites include a high level of variation from individual to individual (polymorphism), an abundant and relatively even distribution throughout the genome, and the ability to be assayed by PCR.
Although the initial genetic-mapping goal has been attained, the 1993 extended 5-year plan recognized the importance of continued improvement in genetic-mapping technology. Easier, automatable, and more cost-effective genotyping methods remain a priority. Such methods probably will require the development of new types of genetic markers, novel genotyping technology, and new analytical tools. Maximizing the usefulness of the genetic map will be particularly important for dissecting the genetics of such complex traits as susceptibilities to heart disease, hypertension, and diabetes.
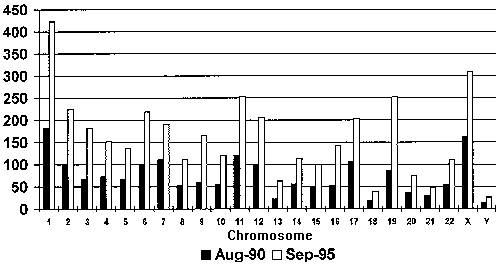
Growth of Mapped Genes, 1990-1995. The number of mapped genes has risen sharply over the past 5 years, from 1772 genes at the inception of the Human Genome Project in 1990 to 3695 genes by September 15, 1995. Gene distribution depicted here may reflect mapping activity per chromosome rather than relative gene density, which will remain unknown until the majority of genes are mapped. Numbers in this graph do not include genes not yet assigned to chromosomes. (Source: GDB, 1995)
PHYSICAL MAPPING
Physical maps are used to isolate and characterize individual genes and other DNA regions of interest and provide the substrate for DNA sequencing. As stated in the 1993 extended 5-year plan [Science 262, 43 46 (1993)], a current Human Genome Project goal for physical mapping is to complete an STS-based map of the human genome with markers spaced every 100 kb on average. Investigators are generating STS maps using both chromosome-specific and genome-wide strategies, and collective progress has been impressive.
Constructing a 100-kb resolution map will require generating and ordering some 30,000 STS markers. A number of different strategies are being applied on a genome-wide basis to build such a map; these strategies include STS-content mapping using large-insert YAC clones, radiation hybrid mapping, and clone fingerprinting. Adoption of a whole-genome approach for map building has been important in the rapid progress of the past 2 to 3 years. Investigators also plan to map a common subset of STS markers on the different maps currently under construction, resulting in well-integrated maps with many more mapped STSs than any one laboratory could produce.
For example, efforts already under way will produce a radiation hybrid map in which a sufficient number of markers will be ordered at very high confidence (1000:1 odds) to provide a resolution higher than 200 kb. Additional STSs will be mapped, albeit with order established at lower confidence levels, with overall map resolution higher than 100 kb. When the map is completed, investigators will be able to select markers from any of the contributing maps, confident that the markers will fall in either the same or adjacent defined regions (or bins) on the chromosome.
Investigators are also placing polymorphic markers within physical maps to allow integration of physical and genetic mapping data across chromosomal regions. These maps will facilitate finer-scale mapping, sequencing, and disease-gene identification. Large-scale efforts to map YACs and BACs onto metaphase chromosomes are linking cytogenetic and sequence/cosmid-based maps.
Initial physical-mapping goals included construction of contig maps (overlapping clone sets) of human chromosomes. Long-range clone contiguity has been achieved for several individual chromosomes in a number of laboratories. Clone-STS maps of entire euchromatic regions of chromosomes 21 and Y were published in 1992. YAC-based clone-STS maps of chromosomes 3, 11, 12, and 22 were finished more recently, and similar maps of chromosomes 4, 5, 7, and X are nearing completion. Maps principally based on cosmid contigs were published recently for chromosomes 16 and 19, and a cosmid-based chromosome 13 map is almost finished.
None of the first-generation physical maps is error free. Errors come from at least two sources: rearrangement of clones relative to the native genome and map-assembly procedures that do not always produce the correct order. Some problems with the initial physical maps will resolve themselves. As marker density increases, internal inconsistencies will become evident and will be corrected upon data reexamination. The use of multiple, independent mapping methods also will contribute significantly to map validation, and using the same markers in different mapping projects will promote quality control. Criteria for assessing and reporting map quality and mapping progress were proposed recently by an international group of scientists.
In spite of these impressive advances, further improvements in mapping technology are essential. New host-vector systems may be required, for example, to capture regions not represented well in current maps and for particular map applications such as sequencing.
DNA SEQUENCING
The most technologically challenging goal of the Human Genome Project remains the complete sequencing of the human genome within the projected 15 years. In the past 5 years, significant progress has been made toward developing the capability for large-scale DNA sequencing. When the genome project began, the longest DNA sequence obtained was the 250,000-bp cytomegalovirus sequence, which took several years to complete. Now, several laboratories have each generated at least 1 Mb; some have determined more than 10 Mb of DNA sequence, mostly from model organisms. The longest contiguous human sequence is 685 kb from the human T-cell Beta receptor locus, a chromosomal region involved in immune responses.
Substantial technical, strategic, and organizational experience in managing large data-production projects has been gained through recent efforts to sequence the genomes of several nonhuman organisms. The capacity of automated sequencing instruments has increased, and newer, higher-throughput instruments are almost ready for introduction into a large-scale sequencing environment. As a result of these and other developments, confidence is growing that continued incremental improvements to current DNA sequencing approaches can be scaled up cost-effectively and probably will enable completion of the first-generation human DNA sequence by 2005.
Continued improvement in sequencing technology will be essential to meet the demands of sequence-based approaches to biological analysis. Achieving the capability for inexpensive sequencing at high-throughput levels will require technology far beyond that available today.
GENE IDENTIFICATION
One of the long-range genome project objectives is to identify all genes and other functional elements in genomic DNA, although understanding their functions will extend far beyond the project. With steady improvements in physical-map resolution and increased sequence data, an attendant need is for robust, high-throughput, and cost-effective methods to identify, map, and study functional elements in the genomes of humans and other organisms.
One method for tabulating genes on a genome-wide basis involves the determination and mapping of unique tags (ESTs) for cDNAs. Identification and initial analysis of large sets of ESTs have been published, and over the next year an even larger number of ESTs are expected to become available. The cDNA clones from which ESTs are derived are also available through the IMAGE Consortium [HGN 6 (6), 3 (March-April 1995)], repositories, and industry. Another international consortium is mapping a large number of publicly available ESTs on both radiation hybrids and YACs. By providing information on the chromosomal locations of genes represented by ESTs, this gene map will increase the value of the EST set for investigators engaged in gene hunting and other analytical activities.
A significant fraction of all human genes is expected to be represented ultimately in the EST and clone sets, but this approach is unlikely to reveal all human genes. Additionally, the amount of sequence and structural information about a gene identified by an EST will be limited. An optimal technology or combination of technologies that will allow high-throughput, cost-effective gene identification remains an important goal of the Human Genome Project.
Disease-Gene Identification
The speed with which human genes are being identified, particularly those responsible for genetic diseases, continues to increase rapidly because of improved genetic and physical maps. As a result, new disease genes are being discovered at a rate of several per month, compared with a few per year not so long ago.
For the past several years, improved maps have increased the efficiency with which investigators use the powerful positional-cloning approach to isolate human disease genes. Positional cloning is essential for identifying genes underlying a particular condition or trait when no prior knowledge of gene function is available.
As genome maps have improved and become increasingly enriched with gene sequences, a new strategy known as positional-candidate cloning has emerged. This approach begins with mapping the disease gene to a small interval on a chromosome. All genes previously identified for that genomic region can then be tested, starting with any whose product suggests possible involvement. Now a gene can become a candidate for disease involvement by virtue of its properties and its map location.
MODEL ORGANISMS
Initial Human Genome Project goals included the characterization of the genomes of such important research organisms as the bacterium Escherichia coli, yeast Saccharomyces cerevisiae, roundworm Caenorhabditis elegans, fruit fly Drosophila melanogaster, and laboratory mouse. These well-studied organisms, which serve as useful, more cost-effective testing grounds for developing large-scale DNA sequencing technology, provide another approach to interpreting human genomic information.
Sequencing
Mouse Maps and Human-Mouse Sequence Comparisons
The mouse genome is about the same size as the human genome. Many genes are conserved between the two species, as is gene order along some chromosomes. Mouse genome maps are thus extremely valuable tools for finding human genes and understanding their functions. This year, investigators completed a genetic map of the mouse genome containing over 6500 microsatellite markers among a total of 7300 genetic markers. Work has begun on a physical map of the mouse genome.
Other investigators are sequencing homologous regions in mouse and human genomes. One example is the region containing the T-cell receptor (TCR) genes that specify cell-surface receptors and play an important role in immune responses. Comparative analysis of this stretch of contiguous sequence from the two species has revealed important and interesting genomic features. These studies are expected to lead to insights into the biological function of TCRs that, in turn, may lead to new ways to counteract transplant rejection, infectious and autoimmune diseases, and allergies.
Other Projects
Human Genome Project sequencing successes have facilitated genome analysis of other interesting and important organisms in the United States and abroad. Examples include the DOE Microbial Genome Initiative for studying organisms of environmental or industrial importance; a privately funded effort that has generated the first complete sequence of the free-living organism Haemophilus influenzae; a project jointly supported by the National Science Foundation, the U.S. Department of Agriculture (USDA), and DOE to map and sequence the genome of the plant Arabidopsis thaliana; and projects focused on mapping the genomes of plants and animals of agricultural importance, organized by USDA and by agencies in other countries.
INFORMATICS
From the beginning of the genome project, informatics has been recognized as essential to the project's success. Much progress has been made in developing computer-based systems for automating the acquisition, management, analysis, and distribution of experimental data. Improvements in laboratory-systems integration and information-management systems have promoted large-scale genomics and other biology programs in academia and industry. A number of new databases have been created, and existing databases have been expanded to allow rapid distribution of genome data. In fact, the number of data sources and programs of interest is too large to summarize in this article, but information about many may be obtained from the NIH and DOE WWW sites.
Improved software is critical to maximizing automated data acquisition and analysis in genetic and physical map construction, base calling, sequence-contig assembly and editing, project management, and feature recognition and annotation.
Beyond the development of these new tools, several other important informatics problems must be solved. The large number of informatics tools and data resources already available or still being developed is not fully integrated and coordinated. Research, development, and coordination efforts are under way to allow easier access to genome research data. With improved computer infrastructure, analyzing information for further and broader biological research will be easier.
Another major challenge is to integrate genome and genome-related databases. Some approaches under discussion include designing common interfaces, implementing "minimonolithic" databases that contain subsets of relevant data extracted from a set of larger public databases, improving database-query tools, and developing a new category of "middleware" to facilitate the construction of federated databases.
Ethical, Legal, and Social Implications
From the outset of the Human Genome Project, researchers recognized that the resulting increase in knowledge about human biology and personal genetic information would raise complex ethical and policy issues for individuals and society. Accordingly, ELSI investigations have been an integral element of genome programs around the world. In the first few years of the U.S. ELSI programs, NIH and DOE have taken two approaches.
The first approach is a research and education grant program supported by 3% to 5% of funds from each agency's budget. The research program has focused on identifying and addressing ethical issues arising from genetic research, responsible clinical integration of new genetic technologies, privacy and the fair use of genetic information, and professional and public education about ELSI issues. Progress in these areas is discussed in separate sections below.
The second approach involves the NIH-DOE Joint Working Group on ELSI of Human Genome Research. This group is charged with exploring and proposing options for sound professional and public policies related to human genome research and its applications and with identifying gaps in the current state of knowledge about ELSI issues.
Ethical Issues Surrounding the Conduct of Genetic Research
The NIH Office of Protection from Research Risks has developed guidelines for protecting the privacy, autonomy, and welfare of individuals and families involved in human genetic research. These recommendations grew out of a series of meetings and studies supported by the NCHGR ELSI program, which has worked with the National Centers for Disease Control and Prevention to develop recommendations for using stored tissue samples in genetic research.
Responsible Clinical Integration of New Genetic Technologies
Rapid development of new testing techniques and DNA-based diagnostic tests raises questions about their appropriate use beyond the research setting. The NCHGR ELSI program has supported a number of studies to identify issues and develop policy recommendations regarding the delivery of genetic tests into clinical practice.
One set of studies examined issues surrounding genetic testing and counseling for cystic fibrosis (CF) mutations. Results from this consortium led to proposals about preferred methods for providing CF testing to those who desire it. On the basis of these and other study results, clinical policy recommendations are expected to emerge from appropriate professional societies.
Last year, a second major effort in introducing genetic tests was initiated with a set of projects to examine testing and counseling for heritable breast, ovarian, and colon cancer risks. Issues include interest in, demand for, and impact of testing as well as alternative ways to provide the service.
In another approach to the use and regulation of new genetic tests, the ELSI Working Group created a Genetic Testing Task Force. This task force is reviewing genetic testing and examining strengths and weaknesses of current practices and policies. If needed, the task force will recommend changes to ensure that only necessary genetic tests are done and that they are conducted by qualified laboratories.
Finally, in 1994, the Institute of Medicine published a study of the clinical integration of new genetic tests. This report offered a number of recommendations for laboratory quality control of DNA diagnostics and for genetic testing in the clinical setting.
Privacy and Fair Use of Genetic Information
Information obtained from genetic testing potentially can serve the individual well by opening the door to therapeutic or preventive intervention. However, this information may also have such unwelcome effects as increased anxiety, altered family relationships, stigmatization, and discrimination on the basis of genotype. Concerns about stigmatization and discrimination are particularly troubling, especially regarding employability and insurability. In 1993 the ELSI Working Group established the Task Force on Genetic Information and Insurance to assess the potential impact of human genetic advances on U.S. health care and to make recommendations for managing that impact within a reformed health-care system.
A Genetic Privacy Act has been drafted with support from the DOE ELSI program. This act, a model for privacy legislation, covers the collection, analysis, storage, and use of DNA samples and the genetic information derived from them. This first legislative product of the ELSI component has been introduced into several state legislatures and was incorporated into a recently passed measure in Oregon. In November, a similar bill was introduced in the U.S. Senate.
Earlier this year, the U.S. Equal Employment Opportunity Commission ruled that genetic discrimination in employment decisions is illegal.
Professional and Public Education
ELSI programs have funded educational projects to increase understanding of the nature and appropriate use of genetic information by health-care professionals, policymakers, and the public. These projects include a reference work to assist federal and state judges in understanding genetic evidence; curriculum modules for middle and high schools; teacher-training workshops; short courses on genome science; radio and television programs on science and ethical issues of the genome project; and the development of educational materials.
REAPING THE BENEFITS
The beginning phase of the Human Genome Project has been remarkably successful. Public data describing human DNA and the DNA of other organisms has expanded enormously, and the information is being used at an increasing rate. Genome project contributions to the study of inherited disease and other biological phenomena are now widely recognized by the scientific community. Investigators are no longer arguing about whether the genome project is a good idea but are debating the most effective ways to reap its rewards. In the commercial sector, a burgeoning body of resources is providing a new base for a wide range of technology industries.
Products of the Human Genome Project including maps, DNA sequences, and improved technology for genomic analysis will soon enable the era of sequence-based biological investigation to begin in earnest.
[Denise Casey, HGMIS, and NCHGR and DOE OHER program staff]
Return to the Table of Contents
The electronic form of the newsletter may be cited in the following style:
Human Genome Program, U.S. Department of Energy, Human Genome News (v7n3).
The Human Genome Project (HGP) was an international 13-year effort, 1990 to 2003. Primary goals were to discover the complete set of human genes and make them accessible for further biological study, and determine the complete sequence of DNA bases in the human genome. See Timeline for more HGP history.

Published from 1989 until 2002, this newsletter facilitated HGP communication, helped prevent duplication of research effort, and informed persons interested in genome research.
