Archive Site Provided for Historical Purposes

Sponsored by the U.S. Department of Energy Human Genome Program
Human Genome News, January-March 1996; 7(5)
Online Mendelian Inheritance in Man (OMIM) is a comprehensive, authoritative, and up-to-date human gene and genetic disorder catalog that supports medical genetics and the Human Genome Project. The print version, Mendelian Inheritance in Man (MIM), was started in the early 1960s by physician Victor McKusick [Johns Hopkins University (JHU)] as a catalog of X-linked traits. The first edition of MIM was printed in 1966 and the 11th in 1994.
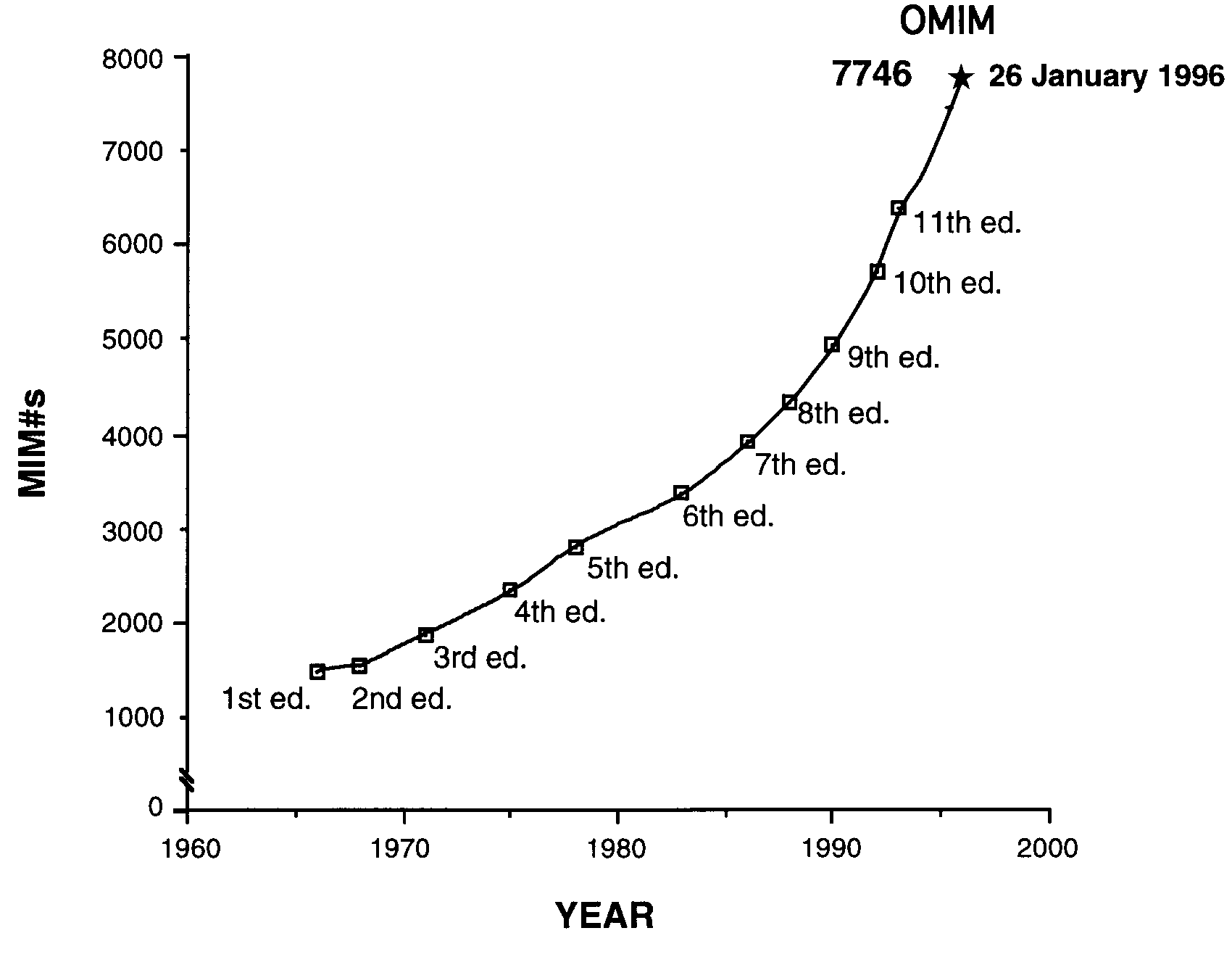
Total number of entries was 7746 on January 26, 1996.
In the early 1980s, MIM was used by an informatics group at the National Library of Medicine (NLM) Lister Hill Center as the testbed in developing the Information Retrieval Experiment (IRx), a system for maintaining and searching full-text knowledge bases. The MIM online version, established in 1985 at the JHU Welch Medical Library, was made publicly available in September 1987 under funding from the Howard Hughes Medical Institute (HHMI). In 1989 HHMI moved the Human Gene Mapping Library, which it also was funding, from New Haven to Baltimore for management and distribution along with OMIM. In 1990 DOE and NIH assumed funding of OMIM and the new entity (Genome Database) as part of the Human Genome Project.
Contents. Early versions of MIM listed Mendelian phenotypes (mainly disorders) classified according to the inheritance mode: autosomal dominant, autosomal recessive, and X-linked. The objective was to create one entry per gene locus. Based mainly on heavily referenced periodical literature, entries described the disorder as well as genetic peculiarities. When phenotype distinctness and inheritance mode were considered quite certain, the entry was honored with an asterisk. The absence of an asterisk indicated uncertainty.
In the 1960s, Mendelian inheritance of a phenotype was almost the only way to define an entry for MIM. In the 1970s, interspecific somatic cell hybrids permitted mapping of genes for which no Mendelian variation had been identified (e.g., thymidine kinase on chromosome 17). These genes of known function but no Mendelian variation were also given entries in MIM.
Since 1980, molecular genetics has permitted the isolation, sequencing, and mapping of many genes and the identification of disease-associated mutations; many have been given entries in OMIM even though no Mendelian phenotype was known.
This evolution was responsible for a change in MIM's subtitle from Catalogs of Autosomal Dominant, Autosomal Recessive, and X-linked Phenotypes (used in the first 10 editions) to Catalogs of Human Genes and Genetic Disorders (used in the 11th edition). The accompanying graph illustrates the growth of entries in MIM (and OMIM) over the more than 30 years of its existence.
Organization. All entries are given a unique six-digit number. Catalogs of Y-linked loci, with ID numbers beginning with 4, and of mitochondrial genes and phenotypes, with ID numbers beginning with 5, were started in 1992. Up until May 15, 1994, separate catalogs were maintained for autosomal dominant, autosomal recessive, and X-linked entries, with ID numbers beginning with 1, 2, and 3, respectively. Any autosomal gene of known function that had been characterized by mapping, cloning, or sequencing but had no known associated Mendelian phenotype or variation was placed in the autosomal dominant catalog.
Autosomal entries created after May 15, 1994, are assigned an arbitrary, consecutive ID number beginning with 6. Thus, in addition to the three chromosome-specific catalogs X, Y, and mitochondrial autosomal loci are now represented also by entries having numbers beginning with 6.
A number sign (#) is used to distinguish some entries that describe a particular phenotype caused by mutations in two or more different genes represented by separate entries.
Special Features. OMIM has two features important to both the science and practice of medical genetics: (1) listing of the mutations (allelic variants) that constitute the molecular basis of genetic diseases and (2) synopsis of the human gene map, with particular attention to the morbid map (i.e., chromosomal sites of genetic disorders).
Allelic variants are identified uniquely by a ten-digit number consisting of the six digits of the primary entry number followed by a dot and four digits beginning with .0001. Thus, the unique entry number for the beta-globin locus (HBB) is 141900 and that for the sickle hemoglobin mutation (HBS) is 141900.0243.
A synopsis of the human gene map has been maintained in the front matter of the print MIM beginning with the third (1971) edition. Users of the online version can move directly from the entry concerning a particular gene or phenotype to the appropriate place in the chromosome-by-chromosome listing of mapped genes, and vice versa. Information on methods and certainty of mapping, disorders due to mutation in the given gene, and mapping of the mouse homolog is also given in tabular form.
Timeliness. OMIM staff members attempt to incorporate journal information as soon as possible. Two-thirds of OMIM references come from 20 "high-impact" journals, some of which distribute embargoed prepublication copies of articles to the lay media.
[NCBI OMIM browser: http://www3.ncbi.nlm.nih.gov/Omim/. NCBI services: info@ncbi.nlm.nih.gov or 301/496-2475.OMIM editorial offices: 410/955-6641, Fax: -4999, mimadm@ncbi.nlm.nih.gov]
Responsibility for distributing OMIM was transferred on December 1, 1995, to the National Center for Biotechnology Information (NCBI) of NLM. OMIM editorial offices and functions will remain at Johns Hopkins Hospital in Baltimore. OMIM formerly was distributed by GDB, which is also at JHU. Moyra Smith, scientific director since March 1, coordinates the distributed multiauthorship. Editorial functions are funded by the NIH National Center for Human Genome Research under a grant to JHU, with David Valle the principal investigator.
GDB will continue to maintain its WWW links to OMIM. NCBI provides WWW, telnet, and dialup access to the IRx version; an e-mail server; and anonymous ftp for downloading files. New OMIM enhancements include direct links to DNA and protein sequence databases and to the Medline database. Most references in OMIM are linked to an abstract in Medline.
Return to the Table of Contents
The electronic form of the newsletter may be cited in the following style:
Human Genome Program, U.S. Department of Energy, Human Genome News (v7n5).
The Human Genome Project (HGP) was an international 13-year effort, 1990 to 2003. Primary goals were to discover the complete set of human genes and make them accessible for further biological study, and determine the complete sequence of DNA bases in the human genome. See Timeline for more HGP history.

Published from 1989 until 2002, this newsletter facilitated HGP communication, helped prevent duplication of research effort, and informed persons interested in genome research.
