Archive Site Provided for Historical Purposes

Sponsored by the U.S. Department of Energy Human Genome Program
In this issue...
In the News
Special Meeting Report
Web, Publications, Resources
Funding Information
Meeting Calendars & Acronyms
Protein function generally is thought to follow from, indeed to require, a specific three-dimensional (3-D) structure. This view arose 100 years ago in Fischers lock-and-key proposal. About 70 years ago Wu and, independently, Mirsky and Pauling proposed that proteins assume particular 3-D structures as the result of weak interactions and that denaturation results from disruption of these weak forces accompanied by loss of specific 3-D structure. This dependence of function on 3-D structure was largely accepted by the time of Anfinsens protein-folding studies. The flood of 3-D structures determined by X-ray diffraction and nuclear magnetic resonance (NMR) has largely drowned out alternative views.
In contrast to the dominant sequence-to-structure-to-function view given above, a few reports on proteins whose functions require disorder* have trickled through the literature for the past 50 years. For example, as early as 1950, Karush provided evidence that serum albumins binding site exists as a structural ensemble with different members in equilibrium with each other. The promiscuity of ligand binding by the albumins is explained by selection of the ensemble member that fits the ligand shape—a process Karush called configurational adaptability.
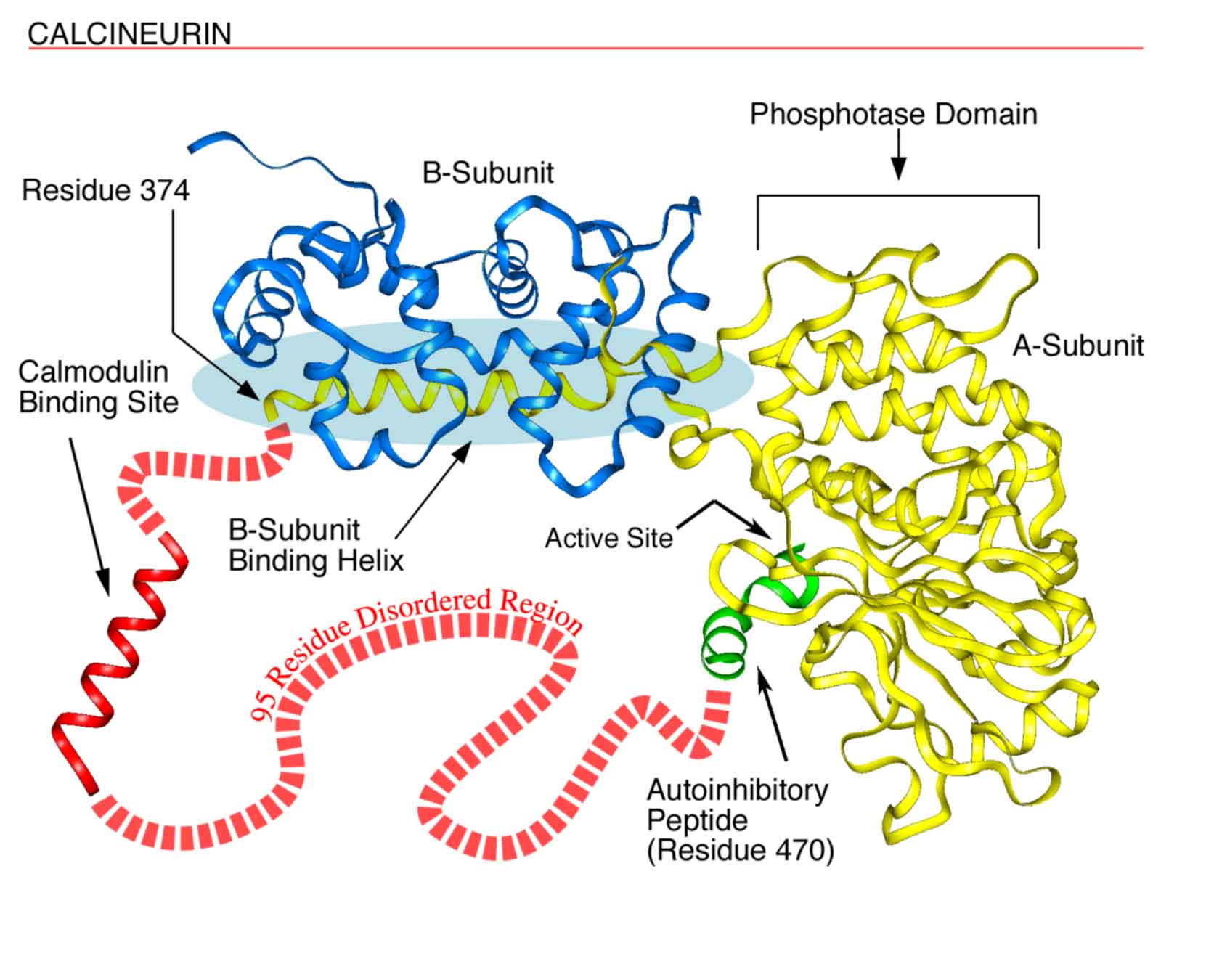
Fig. 1. Disorder in Calcineurin. Calcineurin’s a-subunit contains a globular phosphatase domain, a helical extension that bind the b-subunit, a disordered region not observed in the crystal structure, and an autoinhibitory peptide that binds in the phosphatase domain’s active site. The a-subunit's intrinsically disordered region, containing 95 amino acids, connects the ends of the helical extension (residue 374) and the autoinhibitory peptide (residue 470) and includes a calmodulin binding site. This region probably is disordered at least in part to allow calmodulin to bind. (see Fig. 2).
To provide a more recent example, the calmodulin binding site in calcineurin (Fig. 1) was shown by Klee to be extremely sensitive to protease digestion and thus to be a disordered ensemble; this disorderliness was confirmed in Kissingers X-ray diffraction structure as indicated by missing coordinates in the same region. The disorder is likely to be essential to provide calmodulin (Fig. 2, below) with the space it needs to completely surround its target helix as observed in a calmodulin-target helix cocrystal, the structure of which was determined by Quiocho and colleagues. After these many years, general reviews on intrinsically disordered proteins are just now beginning to appear. In one of these reviews, Wright and Dyson suggested that the existence and commonness of proteins with intrinsic disorder call for a reassessment of the structure-function paradigm.1
In our work we hypothesized that, since amino acid sequence determines 3-D structure, sequence should determine lack of 3-D structure as well. If this were true, the accuracies of disorder predictions using amino acid sequence information would exceed the accuracies expected by chance. From literature and database searches, we collected a set of proteins that were structurally characterized to have regions of disorder under physiological conditions, including a few proteins indicated by NMR to be wholly disordered. Once a set of disordered proteins was assembled, we constructed predictors to test the hypothesis.
For datasets with equal numbers of ordered and disordered residues, our predictors of natural disordered regions (PONDRs) initially were about 70% accurate. The latest PONDR was trained using 16,785 putatively disordered residues from 145 nonhomologous proteins, balanced by an equal number of ordered residues, and gave an accuracy of about 83%.2
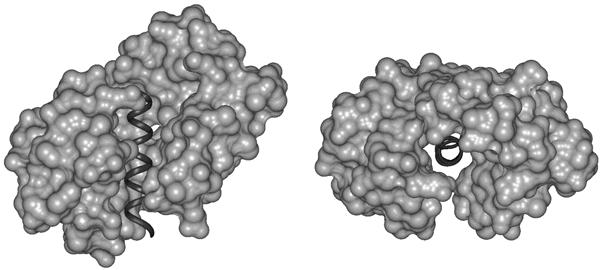
Fig. 2. Disorder Necessary for Calmodulin Binding. Calmodulin (light) bound to the target helix from calmodulin-dependent protein kinase II (dark) is shown in two orientation: (left) from the side and (right) looking down the target helix. Calmodulin completely surrounds the target helix, indicating that calmodulin cannot bind a target helix if the helix is interacting closely with its parent protein.
These accuracies are far above the 50% expected by chance. Thus, the hypothesis that intrinsic disorder is encoded by the sequence is strongly supported. Furthermore, the intrinsically disordered regions have amino acid compositions that are very different from those of ordered proteins in just exactly the way a biochemist would expect. Compared to ordered proteins, disordered proteins are depleted in hydrophobic and, especially, aromatic amino acids. Further, disordered proteins are necessarily enriched in hydrophilic amino acids, often with charge imbalance.
In addition, we have PONDRed the proteomes of more than 30 organisms. The findings were summarized as percentages of the proteins in each proteome predicted to contain long disordered regions (LDRs), where an LDR is a disorder prediction of 40 or more consecutive residues. By this measure, the percentages of proteins with predicted LDRs ranged from 7% to 33% in 22 bacteria, 9% to 37% in 7 archaea, and 36% to 63% in 5 eukaryota. The large jump in LDRs in the multicellular organisms was completely unexpected.
Why such a large jump in LDRs for the eukaryota? We are unsure, but there are some interesting possibilities. We noticed that most of the disordered training examples use their disordered regions for cell signaling or regulation, just as in the calcineurin example cited above. The association between regulatory function or signaling and intrinsic disorder appears, furthermore, to be conserved across all three kingdoms. Qualitatively, it seems reasonable for highly flexible disordered proteins, rather than rigid ones, to be used to respond to environmental changes.
In more detail, Schulz showed that disordered proteins can bind to partners with both high specificity and low affinity because a large fraction of the contact energy has to be used for folding rather than for affinity. Thus, regulatory interactions can be both specific and easily dispersed. This is a major advantage because turning a signal off is as important as turning it on. Karush, Quiocho, and Wright, furthermore, all have pointed out that conformational disorder mediates binding diversity because a flexible chain can adopt different conformations to fit with different ligands. Thus, a significant advantage of intrinsic disorder is to allow one regulatory region or protein to bind to many different partners. The ability to partner with many ligands, potentially including both proteins and nucleic acids, is likely to be of central importance in the development of information networks across the cell membrane as well as inside the cell. Indeed, a recent observation is that the more interactions a given protein makes with other proteins, the more likely that a deletion will lead to lethality.3
While attempting to organize our thoughts about the various relationships between intrinsic disorder and protein function, we created the Protein Trinity Hypothesis (Fig. 3). In this view, native proteins can be in one of three states: the solid-like ordered state, the liquid-like collapsed-disordered state, or the gas-like extended-disordered state. Function is then viewed to arise from any one of the three states or from transitions among them.
*Disordered regions are amino acid sequences within proteins that fail to fold into a fixed structure and are involved in a variety of biological functions. Back
References
Keith Dunker, Washington State University, and Zoran Obradovic, Temple University
The electronic form of the newsletter may be cited in the following style:
Human Genome Program, U.S. Department of Energy, Human Genome News (v12n1-2).
The Human Genome Project (HGP) was an international 13-year effort, 1990 to 2003. Primary goals were to discover the complete set of human genes and make them accessible for further biological study, and determine the complete sequence of DNA bases in the human genome. See Timeline for more HGP history.

Published from 1989 until 2002, this newsletter facilitated HGP communication, helped prevent duplication of research effort, and informed persons interested in genome research.