It is important that any theoretical improvements in minimisation procedures are tested in actual calculations. In order to test the different procedures at an affordable computational cost, a non-interacting test system was selected. In a non-interacting system the exact wavefunction for the system is known, which helps in the analysis of minimisation procedures. We believe that the conclusions drawn from this systems are general because in more realistic calculations the problems simply become more serious.
The model system consistent of an fcc simulation cell containing 16 atoms and
64-electrons. The electrons were subject to a local potential which
is described by two Fourier components, ![]() = -0.1 a.u., and
= -0.1 a.u., and
![]() = -0.06 a.u., chosen to give a reasonable description of the
valence bandstructure of silicon.
These values are in reasonable agreement with empirical
pseudopotential form factors for silicon [85]. Overall
this model gives a reasonable description of the valence states of
silicon and retains the essential features for testing the
optimization techniques.
= -0.06 a.u., chosen to give a reasonable description of the
valence bandstructure of silicon.
These values are in reasonable agreement with empirical
pseudopotential form factors for silicon [85]. Overall
this model gives a reasonable description of the valence states of
silicon and retains the essential features for testing the
optimization techniques.
Near exact single-particle orbitals were obtained by diagonalising
the Hamiltonian in a plane-wave basis set containing all waves up to
a kinetic energy cutoff of 15 a.u. This basis set is still
incomplete, but the square root of the variance of the energy is about
0.02 eV per atom, which is negligible for current purposes. A
variational parameter, ![]() , was added to the wavefunction in the
form of a
, was added to the wavefunction in the
form of a ![]() function with the full symmetry of the diamond
structure:
function with the full symmetry of the diamond
structure:
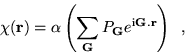 |
(5.10) |
where ![]() labels the 8 reciprocal lattice vectors of
the [111] star and
labels the 8 reciprocal lattice vectors of
the [111] star and ![]() is a phase factor associated with the
non-symmorphic symmetry operations. The many-body wavefunction is
given by
is a phase factor associated with the
non-symmorphic symmetry operations. The many-body wavefunction is
given by
![\begin{displaymath}
\psi=D^{\uparrow}D^{\downarrow}
\exp\left[ \sum_{i=1}^{N} \chi({\bf r}_{i})\right] \;\;\;.
\end{displaymath}](pkthimg529.gif) |
(5.11) |
The exact value of the parameter, ![]() , is, of course, zero. To
model the situation where the wavefunction does not possess the
variational freedom to encompass the exact one we used a smaller basis
set cutoff of 2.5 a.u. The variational energy from this wavefunction
is 0.35 eV per atom above the exact value, which is typical of the
values we encounter in our solid state calculations. The optimal
value of
, is, of course, zero. To
model the situation where the wavefunction does not possess the
variational freedom to encompass the exact one we used a smaller basis
set cutoff of 2.5 a.u. The variational energy from this wavefunction
is 0.35 eV per atom above the exact value, which is typical of the
values we encounter in our solid state calculations. The optimal
value of ![]() for this inexact wavefunction is very close to zero.
for this inexact wavefunction is very close to zero.
This model exhibits all the numerical problems encountered in optimization procedures. In practical situations one may have more electrons and more parameters to optimize, which makes the numerical instabilities more pronounced. In order to analyse the behaviour in detail the variance of the difference objective functions was analysed. Unfeasibly large numbers of electron configurations were required to obtain accurate values of the variance of the objective functions for wavefunctions with more electrons and variable parameters than used in the model system. For these systems, improvements in optimisation procedures will be even more advantageous.
A sample of
![]() statistically independent
electronic configurations was generated and used to calculate the quantities
involved in the various optimization schemes. In practical
QMC applications, a typical number of configurations used for
optimisation might be
statistically independent
electronic configurations was generated and used to calculate the quantities
involved in the various optimization schemes. In practical
QMC applications, a typical number of configurations used for
optimisation might be ![]() ,
but it was necessary to use a much larger number in these tests to obtain
sufficiently accurate values of the different objective functions and,
in particular, their variances. In a practical application an
objective function, say
,
but it was necessary to use a much larger number in these tests to obtain
sufficiently accurate values of the different objective functions and,
in particular, their variances. In a practical application an
objective function, say ![]() , is evaluated using, say,
, is evaluated using, say, ![]() configurations. The quantities of interest are then
configurations. The quantities of interest are then ![]() and
its variance calculated as averages over blocks of
and
its variance calculated as averages over blocks of ![]() configurations.
configurations.
The configurations were generated by a Metropolis walk distributed
according to ![]() , using the (inexact) reduced basis-set wave
function. An optimization procedure typically starts with
non-optimal parameter values which are improved during the
optimization procedure. Results are presented for configurations
generated with the non-optimal value of
, using the (inexact) reduced basis-set wave
function. An optimization procedure typically starts with
non-optimal parameter values which are improved during the
optimization procedure. Results are presented for configurations
generated with the non-optimal value of ![]() = 0.03, which
gives results typical of the starting value for an optimization, and
= 0.03, which
gives results typical of the starting value for an optimization, and
![]() = 0, which is the final value from a successful
optimization procedure. The qualitative behaviour was found to be not
strongly influenced by the value of
= 0, which is the final value from a successful
optimization procedure. The qualitative behaviour was found to be not
strongly influenced by the value of ![]() .
.
The unweighted mean energy has a maximum at ![]() , i.e.,
the value from which the configurations were generated. This result
can be understood as follows. Consider a wave function of the form
, i.e.,
the value from which the configurations were generated. This result
can be understood as follows. Consider a wave function of the form
where the ![]() are parameters, the
are parameters, the ![]() are
correlation functions and the
are
correlation functions and the ![]() ,
,
![]() are up
and down-spin determinants. The mean unweighted energy (equation
5.7) can be written as
are up
and down-spin determinants. The mean unweighted energy (equation
5.7) can be written as
where ![]() are the parameter values from which the
configurations are generated and
are the parameter values from which the
configurations are generated and
| (5.14) |
The ![]() and the constant term depend on the
and the constant term depend on the
![]() but not on the
but not on the ![]() . When there is only a single
parameter,
. When there is only a single
parameter, ![]() is negative, so that
is negative, so that ![]() is a quadratic function with
a maximum at
is a quadratic function with
a maximum at
![]() . When there is more than one
parameter the stationary point of the quadratic can be a maximum,
minimum or saddle point, which is not acceptable behaviour for an
objective function. The weights may be altered in other ways, such as
limiting their upper value, but if the weights are altered the minima
of the energy are moved, which is a ``strong bias'' in the objective
function. Therefore, if an energy minimisation method is used,
weights must be included.
. When there is more than one
parameter the stationary point of the quadratic can be a maximum,
minimum or saddle point, which is not acceptable behaviour for an
objective function. The weights may be altered in other ways, such as
limiting their upper value, but if the weights are altered the minima
of the energy are moved, which is a ``strong bias'' in the objective
function. Therefore, if an energy minimisation method is used,
weights must be included.
![\includegraphics [width=10cm]{Figures/varmin_fig1a.eps}](pkthimg543.gif)
![\includegraphics [width=10cm]{Figures/varmin_fig1b.eps}](pkthimg544.gif)
|
The distributions of the weights and local energies of the test
ensemble are now examined. In figure 5.2 the distributions
of the weights for ![]() = 0.03 and
= 0.03 and ![]() = 0 and for
= 0 and for
![]() = 0 and
= 0 and ![]() = 0.03 are plotted, while in
figure 5.3 the corresponding distributions of the local
energies are shown. The distributions of the weights resemble Poisson
distributions, but the square roots of the variances are significantly
greater than the means, so there are more configurations at large
weights than for a Poisson distribution with the same mean. The local
energies follow normal distributions relatively well. As expected,
the distributions of the local energies is wider for the
= 0.03 are plotted, while in
figure 5.3 the corresponding distributions of the local
energies are shown. The distributions of the weights resemble Poisson
distributions, but the square roots of the variances are significantly
greater than the means, so there are more configurations at large
weights than for a Poisson distribution with the same mean. The local
energies follow normal distributions relatively well. As expected,
the distributions of the local energies is wider for the ![]() =
0.03 wavefunction. Closer inspection reveals that the distribution of
the local energies is not exactly normal because the actual
distributions have ``fat tails''. The outlying energies result from
outlying values of the kinetic energy. The standard deviations are
=
0.03 wavefunction. Closer inspection reveals that the distribution of
the local energies is not exactly normal because the actual
distributions have ``fat tails''. The outlying energies result from
outlying values of the kinetic energy. The standard deviations are
![]() = 0.964 a.u. and
= 0.964 a.u. and ![]() = 0.726 a.u., for
= 0.726 a.u., for ![]() = 0.03
and 0, respectively. The expected percentage of configurations beyond
3
= 0.03
and 0, respectively. The expected percentage of configurations beyond
3![]() from the mean of a normal distribution is 0.27%, but the
actual percentages are 0.443% and 0.608% for
from the mean of a normal distribution is 0.27%, but the
actual percentages are 0.443% and 0.608% for ![]() = 0.03 and
0, respectively. Although these outlying local energies give a
negligible contribution to the mean energy, calculated with or without
weighting, and only a very small contribution to the values of the
variance-like objective functions,
= 0.03 and
0, respectively. Although these outlying local energies give a
negligible contribution to the mean energy, calculated with or without
weighting, and only a very small contribution to the values of the
variance-like objective functions, ![]() ,
, ![]() , and
, and
![]() , they give significant contributions to the variances of
the variance-like objective functions.
, they give significant contributions to the variances of
the variance-like objective functions.
![\includegraphics [width=10cm]{Figures/varmin_fig2.eps}](pkthimg546.gif)
|
![\begin{displaymath}
\Phi = D^{\uparrow}D^{\downarrow} \exp \left[\sum_k \alpha_k J_k \right] \;\;,
\end{displaymath}](pkthimg533.gif)
![\includegraphics [width=10cm]{Figures/varmin_fig3.eps}](pkthimg547.gif)