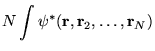The one-body density matrix is an object fundamental to
quantum mechanics. Its complete knowledge enables the computation of
all one-body expectation values without reference to a system's
wavefunction. The one-body density
matrix [109,110,111] for a
normalized wave function, ![]() , is defined as
, is defined as
The pair-correlation function of the valence electrons of bulk silicon, which is related to components of the two-body density matrix, was recently computed using VMC. [14] Substantial inhomogeneity and deviations from single particle theories were found, suggesting that other distribution functions, such as the one-body density matrix should be examined. No previous studies of the entire one-body density matrix using accurate many-body wavefunctions were found in the literature.
Although the one-body density matrix is of immediate interest, the matrix also provides a potential route to wavefunction optimisation and to the computation of excitation energies. In this chapter, a QMC scheme for sampling the one-body density matrix is developed and applied to the valence electrons of bulk silicon. The natural orbitals are obtained by diagonalising the density matrix and tested for improvements in the DMC fixed-node energy. The relation between the density matrix, the momentum density and Compton profiles is briefly discussed, and the zero momentum component of the momentum density computed. Approximate excitation energies are also obtained, via an extension of Koopmans' theorem, [112] again with the density matrix as a key quantity.
The question of which single-particle orbitals lead to the best approximation to the exact many-body wave function is still open. Furthermore, this choice fixes the nodal surface of the trial wave function and therefore determines the accuracy of the fixed-node approximation. LDA and HF orbitals have been used successfully in a number of atomic,[46,37] molecular[113,114] and solid[115] QMC calculations, but so far it has not proved possible to perform a direct optimization of the single-particle orbitals of an extended system. A study of first row atoms and molecules[116,53] showed that lower energies can be obtained in both VMC and DMC using a trial wave function containing several determinants obtained from a multi-configuration self-consistent field (MCSCF) calculation. However, a similar study for small silicon clusters [45] found that trial wave functions containing a single determinant of natural orbitals computed within an MCSCF scheme gave better DMC results than some multi-determinant wave functions. This result strongly suggests that the natural orbitals result in improved nodal surfaces, and motivates the calculation of the natural orbitals for bulk silicon.
An expansion of a wave function in Slater determinants of natural orbitals requires a smaller number of terms for a given accuracy than expansions using other orbitals.[2,4] Calculation of the natural orbitals is, however, costly, and less expensive schemes such as natural pair orbitals[117,118] have been proposed to improve convergence in quantum chemical calculations. It is not clear that orbitals arising in schemes designed to accelerate convergence of configuration interaction (CI) calculations should give smaller fixed-node errors in DMC calculations than LDA or HF orbitals. Although a CI wavefunction can well approximate an exact eigenstate, whether the low order terms in the expansion improve the nodes in general is not known. However, as mentioned above, there is some evidence to suggest that natural orbitals have this property. Natural orbitals have not frequently been computed within fermion QMC, although VMC and DMC calculations of natural orbitals have been reported for the ground states of the Li, C, and Ne atoms.[67] No calculations of natural orbitals for realistic extended fermion systems using many-body wavefunctions have appeared in the literature prior to this study, although for homogeneous systems the translational symmetry requires the natural orbitals to be plane waves.
Systematic studies of multi-determinant wave functions in QMC are lacking for solids. It seems reasonable to assume that multi-determinant wave functions will have improved nodes, and therefore give a better representation of the exact wave function, but there is little direct evidence to support this. Multi-configurational approaches include correlation effects, but do so relatively inefficiently - large numbers of terms (configurations) are usually required to obtain a significant proportion of the correlation energy. This form of wave function is unattractive for QMC as a representation of the wave function which is both accurate and can be evaluated rapidly is required. By obtaining the one-body density matrix and hence the natural orbitals from a VMC calculation using a correlated trial wave function, the need to determine them using a multi-determinantal calculation is bypassed.
QMC techniques may be applied to excited states, and consequently there is considerable interest in calculating excitation energies and optical properties of materials. Excitation energies may be obtained by analyzing DMC decay curves,[119,120] but this method has not proven very useful due to the large statistical noise. Furthermore, as the quality of the ground state trial wave function improves, less information about excited states is obtained. A combination of ground and excited state wave functions must then be used to obtain upper bounds for the excitation energies.
Direct methods for calculating excitation energies have met with more success. Mitás and Martin have calculated an excitation energy in a molecular nitrogen solid by performing DMC calculations for the ground and excited states.[42] Mitás has also reported similar calculations for two excitation energies in diamond.[101] Recently[103] 27 excitation energies of silicon were calculated using DMC, obtaining very good agreement with experiment for the low lying excitation energies, while the energies of the higher lying excitations were somewhat too large. Although successful, these techniques are computationally very expensive. The energy of each state of interest is calculated separately and excitation energies computed as energy differences.
A different approach to the direct methods is taken here, using the ``Extended Koopmans' Theorem'' (EKT).[98,99] The theorem derives from quantum chemistry and involves the one-body density matrix. The EKT has not previously been applied to extended systems, or applied to explicitly many-body wavefunctions.
The EKT may be approximated by a simplified diagonal approximation to the full theory. This approximation was used previously to estimate quasihole energies in silicon[121] and NiO.[122] The EKT and its simplified form require only a single ground state calculation in contrast to direct techniques, allowing more efficient studies of excitations in large systems. If sufficiently accurate the EKT would clearly be a significant advance over direct techniques.