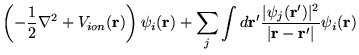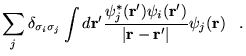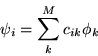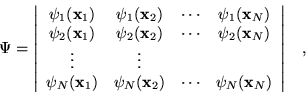 |
(2.4) |
where the variables ![]() include the coordinates of space and
spin. This simple ansatz for the wavefunction
include the coordinates of space and
spin. This simple ansatz for the wavefunction ![]() captures much of the
physics required for accurate solutions of the Hamiltonian. Most
importantly, the wavefunction is antisymmetric with respect to an
interchange of any two electron positions. This property is required
by the Pauli exclusion principle, i.e.
captures much of the
physics required for accurate solutions of the Hamiltonian. Most
importantly, the wavefunction is antisymmetric with respect to an
interchange of any two electron positions. This property is required
by the Pauli exclusion principle, i.e.
| (2.5) |
This wavefunction may be inserted into the Hamiltonian, equation
2.3, and an expression for the total energy
derived. [2,3,4] Applying
the theorem that the value of a determinant is unchanged by any
non-singular linear transformation, we may choose the ![]() to be an
orthonormal set. We now introduce a Lagrange multiplier
to be an
orthonormal set. We now introduce a Lagrange multiplier ![]() to
impose the condition that the
to
impose the condition that the ![]() are normalised, and minimise with
respect to the
are normalised, and minimise with
respect to the ![]()
![\begin{displaymath}
\frac{\delta}{\delta\psi}\left[<\hat{H}>-\sum_j\epsilon_j\int\vert\psi_j\vert^2d{\bf r}\right] = 0 \;\;.
\end{displaymath}](pkthimg37.gif) |
(2.6) |
An enormous simplification of the expressions for the orbitals ![]() results. They reduce to a set of one-electron equations of the form
results. They reduce to a set of one-electron equations of the form
| (2.7) |
where ![]() is a non-local potential and the local ionic
potential is denoted by
is a non-local potential and the local ionic
potential is denoted by ![]() . The one-electron equations resemble
single-particle Schrödinger equations.
. The one-electron equations resemble
single-particle Schrödinger equations.
The full Hartree-Fock equations are given by
The right hand side of the equations consists of four terms. The first
and second give rise are the kinetic energy contribution and the
electron-ion potential. The third term, or Hartree term, is the simply
electrostatic potential arising from the charge distribution of ![]() electrons. As written, the term includes an unphysical
self-interaction of electrons when
electrons. As written, the term includes an unphysical
self-interaction of electrons when ![]() . This term is cancelled in
the fourth, or exchange term. The exchange term results from our
inclusion of the Pauli principle and the assumed determinantal form of
the wavefunction. The effect of exchange is for electrons of like-spin
to avoid each other. Each electron of a given spin is consequently
surrounded by an ``exchange hole'', a small volume around the electron
which like-spin electron avoid.
. This term is cancelled in
the fourth, or exchange term. The exchange term results from our
inclusion of the Pauli principle and the assumed determinantal form of
the wavefunction. The effect of exchange is for electrons of like-spin
to avoid each other. Each electron of a given spin is consequently
surrounded by an ``exchange hole'', a small volume around the electron
which like-spin electron avoid.
The Hartree-Fock approximation corresponds to the
conventional single-electron picture of electronic structure: the
distribution of the ![]() electrons is given simply by the sum of
one-electron distributions
electrons is given simply by the sum of
one-electron distributions ![]() . This allows concepts such as
labelling of electrons by angular momenta (`` a 3d electron in a
transition metal''), but it must be remembered that this is an
artifact of the initial ansatz and that in some systems modifications
are required to these ideas.
. This allows concepts such as
labelling of electrons by angular momenta (`` a 3d electron in a
transition metal''), but it must be remembered that this is an
artifact of the initial ansatz and that in some systems modifications
are required to these ideas.
Hartree-Fock theory, by assuming a single-determinant form for the wavefunction, neglects correlation between electrons. The electrons are subject to an average non-local potential arising from the other electrons, which can lead to a poor description of the electronic structure. Although qualitatively correct in many materials and compounds, Hartree-Fock theory is insufficiently accurate to make accurate quantitative predictions.
In practice, a basis of plane waves in periodic systems or localised
Gaussians in finite systems, is commonly used. The basis set
expansion represents an additional limitation of the techniques:
unless the basis set expansion has sufficient freedom to encompass the
exact solutions for the Hartree-Fock orbitals, ![]() , a compromise
solution with a higher Hartree-Fock energy will be found2.1. In practical applications, convergence of the basis set must
be studied to verify that the expansion is sufficiently complete.
, a compromise
solution with a higher Hartree-Fock energy will be found2.1. In practical applications, convergence of the basis set must
be studied to verify that the expansion is sufficiently complete.