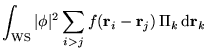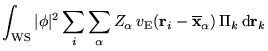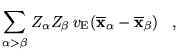In this section the CFSE is considered and the theory of the MPC interaction is described. These effects do not occur in single-particle theories and are more difficult to compensate for than the IPFSE.
The origin of the CFSE was described in section 6.4 as
arising from the XC energy and the dependence of the Ewald
interaction, ![]() , on the size and shape of the simulation
cell. Expanding
, on the size and shape of the simulation
cell. Expanding ![]() around zero separation
gives [86,87]
around zero separation
gives [86,87]
where ![]() is the volume of the simulation cell, and the
tensor D depends on the shape of the simulation cell. (For a
cubic cell D= I.) The constant term arises from the
condition that the average of
is the volume of the simulation cell, and the
tensor D depends on the shape of the simulation cell. (For a
cubic cell D= I.) The constant term arises from the
condition that the average of ![]() over the simulation cell is
zero. The term of order
over the simulation cell is
zero. The term of order ![]() and the higher order deviations from
and the higher order deviations from
![]() make the Ewald interaction periodic and ensure that the sum of
interactions between the particles in the simulation cell gives the
potential energy per cell of an infinite periodic lattice. These
terms are responsible for the spurious contribution to the XC energy,
which is the source of the large finite size effect in many-body
calculations using the Ewald
interaction. [86,87] For cubic
cells the interaction at short distances is larger than
make the Ewald interaction periodic and ensure that the sum of
interactions between the particles in the simulation cell gives the
potential energy per cell of an infinite periodic lattice. These
terms are responsible for the spurious contribution to the XC energy,
which is the source of the large finite size effect in many-body
calculations using the Ewald
interaction. [86,87] For cubic
cells the interaction at short distances is larger than ![]() and
therefore the XC energy is more negative than it should be, and
because the leading order correction is proportional to the inverse of
the simulation cell volume the error per electron is inversely
proportional to the number of electrons in the cell.
and
therefore the XC energy is more negative than it should be, and
because the leading order correction is proportional to the inverse of
the simulation cell volume the error per electron is inversely
proportional to the number of electrons in the cell.
Clearly it is desirable to remove this spurious contribution to the XC
energy, but the Hartree energy is correctly evaluated using the Ewald
interaction. The key requirements for a model Coulomb interaction
giving small CFSEs in simulations with periodic boundary conditions
are therefore: (i) it should give the Ewald interaction for the
Hartree terms, and (ii) it should be exactly ![]() for the interaction
with the XC hole. Unfortunately, the only periodic solution of
Poisson's equation for a periodic array of charges is the Ewald
interaction, which obeys criterion (ii) only in the limit of an
infinitely large simulation cell. The ``Model Periodic Coulomb''
(MPC) interaction satisfies both criteria, but does not satisfy
Poisson's equation.
for the interaction
with the XC hole. Unfortunately, the only periodic solution of
Poisson's equation for a periodic array of charges is the Ewald
interaction, which obeys criterion (ii) only in the limit of an
infinitely large simulation cell. The ``Model Periodic Coulomb''
(MPC) interaction satisfies both criteria, but does not satisfy
Poisson's equation.
The MPC interaction can be written as [87]
The definition of the cutoff Coulomb function, ![]() , involves
a minimum image convention whereby the inter-electron distance,
, involves
a minimum image convention whereby the inter-electron distance, ![]() , is reduced into the Wigner-Seitz (WS) cell of the simulation
cell lattice by removal of simulation cell lattice translation
vectors, leaving a vector
, is reduced into the Wigner-Seitz (WS) cell of the simulation
cell lattice by removal of simulation cell lattice translation
vectors, leaving a vector
![]() . This ensures that
. This ensures that
![]() has the correct translational and point group
symmetry. The first term in equation 6.8 is a direct
Coulomb interaction between electrons within the simulation cell and
the second term is a sum of potentials due to electrons ``outside the
simulation cell''. Note that the second term is a one-body potential
similar to the Hartree potential. It depends on the electronic charge
density,
has the correct translational and point group
symmetry. The first term in equation 6.8 is a direct
Coulomb interaction between electrons within the simulation cell and
the second term is a sum of potentials due to electrons ``outside the
simulation cell''. Note that the second term is a one-body potential
similar to the Hartree potential. It depends on the electronic charge
density, ![]() , but is not a function of inter-particle separation.
, but is not a function of inter-particle separation.
The electron-electron contribution to the total energy is evaluated
as the expectation value of
![]() with the many-electron
wave function,
with the many-electron
wave function, ![]() , minus a double counting term:
, minus a double counting term:
Evaluating the expectation value gives
where the first term on the right hand side is the Hartree
energy and the term in brackets is the XC energy. We can see
immediately that the Hartree energy is calculated with the Ewald
interaction while the XC energy (expressed as the difference between a
full Coulomb term and a Hartree term) is calculated with the cutoff
interaction ![]() .
.
The charge density ![]() appears in equations 6.8,
6.10, and 6.11. In QMC methods, the charge
density is known with greatest statistical accuracy at the end of the
calculation. This is not a serious complication for VMC simulations
as the interaction energy may be evaluated at the end of the
simulation using the accumulated charge density. In DMC this is not
possible because the local energy is required at every step. This
point is investigated in section 6.8.3.
appears in equations 6.8,
6.10, and 6.11. In QMC methods, the charge
density is known with greatest statistical accuracy at the end of the
calculation. This is not a serious complication for VMC simulations
as the interaction energy may be evaluated at the end of the
simulation using the accumulated charge density. In DMC this is not
possible because the local energy is required at every step. This
point is investigated in section 6.8.3.
Consider a simulation cell with periodic boundary conditions
containing ![]() electrons at positions
electrons at positions ![]() and
and ![]() nuclei of
charge
nuclei of
charge ![]() at positions
at positions
![]() . The wave
function of the electrons and nuclei is
. The wave
function of the electrons and nuclei is
![]() , and the total charge density (electrons and nuclei)
is
, and the total charge density (electrons and nuclei)
is
![]() . The interaction energy calculated with
the MPC interaction is
. The interaction energy calculated with
the MPC interaction is
We now employ the adiabatic approximation to separate the electronic and nuclear dynamical variables:
where the
![]() appear only as parameters
in
appear only as parameters
in ![]() . To make further progress a form for the nuclear part of
the wave function,
. To make further progress a form for the nuclear part of
the wave function, ![]() , must be assumed. The simplest assumption
is that
, must be assumed. The simplest assumption
is that ![]() can be written as an appropriately symmetrized product
of very strongly localized non-overlapping single-nucleus functions.
Regardless of whether the product is antisymmetrized, symmetrized, or
not symmetrized, equation 6.12 reduces to
can be written as an appropriately symmetrized product
of very strongly localized non-overlapping single-nucleus functions.
Regardless of whether the product is antisymmetrized, symmetrized, or
not symmetrized, equation 6.12 reduces to
where the
![]() denote the centers
of the single-nucleus functions and
denote the centers
of the single-nucleus functions and ![]() is the electron density.
Note that the electron-nucleus and nucleus-nucleus terms involve
only the Ewald interaction and that the first two terms of
equation 6.14 correspond precisely to the
electron-electron interaction of equation 6.11. This result
justifies the use of equation 6.8 for the
electron-electron interactions while retaining the Ewald interaction
for the electron-nucleus and nucleus-nucleus terms.
is the electron density.
Note that the electron-nucleus and nucleus-nucleus terms involve
only the Ewald interaction and that the first two terms of
equation 6.14 correspond precisely to the
electron-electron interaction of equation 6.11. This result
justifies the use of equation 6.8 for the
electron-electron interactions while retaining the Ewald interaction
for the electron-nucleus and nucleus-nucleus terms.
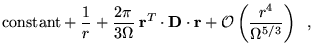
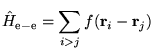
![$\displaystyle \sum_{i}\int_{\rm WS} \left[ v_{{\rm E}}({\bf r}_i-{\bf r})- f({\bf r}_i-{\bf r})\right] {\rho({\bf r})} \, {\rm d}{\bf r} \;\;,$](pkthimg593.gif)
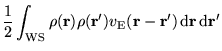
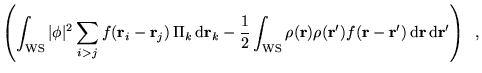
![$\displaystyle \frac{1}{2}\int_{\rm WS} \rho_{\rm T}({\bf r})\rho_{\rm T}({\bf r...
...}({\bf r}-{\bf r}') - f({\bf r}-{\bf r}')] \, {\rm d}{\bf r} \, {\rm d}{\bf r}'$](pkthimg606.gif)
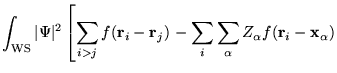
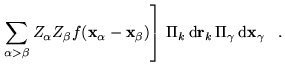
![$\displaystyle \frac{1}{2}\int_{\rm WS} {\rho} ({\bf r}){\rho}
({\bf r}') [v_{{\...
...}({\bf r}-{\bf r}') - f({\bf r}-{\bf r}')] \,
{\rm d}{\bf r} \, {\rm d}{\bf r}'$](pkthimg611.gif)