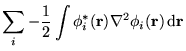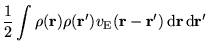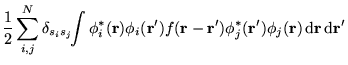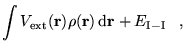The MPC interaction was tested by performing calculations using HF
theory, VMC and DMC. LDA-DFT was used for the IPFSE corrections. A
series of calculations on diamond structure silicon using fcc
simulation cells of increasing size. Cells whose translation vectors are
![]() times those of the primitive unit cell were used.
times those of the primitive unit cell were used.
In an initial set of tests LDA and HF results were compared. These
tests are inexpensive and are not subject to statistical errors
because Monte Carlo techniques are not involved. Simulation cells
with ![]() = 1, 2, 3, 4, and 5, which contain 2, 16, 54, 128, and 250
ions, respectively were considered. The Si
= 1, 2, 3, 4, and 5, which contain 2, 16, 54, 128, and 250
ions, respectively were considered. The Si![]() ions were
represented by norm-conserving non-local LDA pseudopotentials and
the calculations were performed using a plane-wave basis set with a
cutoff energy of 15 Ry.
ions were
represented by norm-conserving non-local LDA pseudopotentials and
the calculations were performed using a plane-wave basis set with a
cutoff energy of 15 Ry.
To measure the IPFSE and the effect of k-point selection,
calculations were performed using
![]() and
and
![]() . To facilitate comparison the HF energy was evaluated
non-consistently with the LDA orbitals, so that the energy differences
arise solely from the difference between the LDA XC energy and the HF
exchange energy.
. To facilitate comparison the HF energy was evaluated
non-consistently with the LDA orbitals, so that the energy differences
arise solely from the difference between the LDA XC energy and the HF
exchange energy.
The HF energy evaluated with the MPC interaction is
where ![]() is the ion-ion energy calculated with
the Ewald interaction. Note that the Hartree energy is evaluated with
the Ewald interaction while the exchange energy is evaluated with
is the ion-ion energy calculated with
the Ewald interaction. Note that the Hartree energy is evaluated with
the Ewald interaction while the exchange energy is evaluated with ![]() ,
the cutoff Coulomb interaction.
,
the cutoff Coulomb interaction.
In figure 6.1 the deviations of the LDA and HF energies
from the fully converged values as a function of system size for
![]() wave functions are shown. The LDA energy converges
smoothly with system size but for small system sizes the IPFSE error
is large because of the
wave functions are shown. The LDA energy converges
smoothly with system size but for small system sizes the IPFSE error
is large because of the ![]() -point sampling. HF energies
calculated with the Ewald and MPC interactions, with and without
incorporating the IPFSE corrections obtained from the LDA data are
shown. The data incorporating the IPFSE (filled symbols) show the
residual CFSE errors. The IPFSE is positive while the CFSE is
negative, in accord with the analysis of the CFSE given in
section 6.7. The IPFSE corrected data show that the CFSE
for the MPC interaction is roughly half that for the Ewald
interaction.
-point sampling. HF energies
calculated with the Ewald and MPC interactions, with and without
incorporating the IPFSE corrections obtained from the LDA data are
shown. The data incorporating the IPFSE (filled symbols) show the
residual CFSE errors. The IPFSE is positive while the CFSE is
negative, in accord with the analysis of the CFSE given in
section 6.7. The IPFSE corrected data show that the CFSE
for the MPC interaction is roughly half that for the Ewald
interaction.
In figure 6.2 similar data for
![]() is
shown. The LDA energy converges rapidly and smoothly with system
size, and therefore the IPFSE is small, which demonstrates the
efficacy of the ``special k-points'' method. The IPFSE and CFSE
errors tend to cancel and for
is
shown. The LDA energy converges rapidly and smoothly with system
size, and therefore the IPFSE is small, which demonstrates the
efficacy of the ``special k-points'' method. The IPFSE and CFSE
errors tend to cancel and for
![]() sampling the HF
data converge more rapidly without the IPFSE corrections. For
sampling the HF
data converge more rapidly without the IPFSE corrections. For ![]() = 3,
corresponding to a 54 atom simulation cell, the LDA finite size error
(IPFSE only) is 0.011 eV per atom, which is very much smaller than the
HF (Ewald) finite size error of -0.211 eV per atom or the equivalent
HF (MPC) error of -0.071 eV per atom. As in the case of
= 3,
corresponding to a 54 atom simulation cell, the LDA finite size error
(IPFSE only) is 0.011 eV per atom, which is very much smaller than the
HF (Ewald) finite size error of -0.211 eV per atom or the equivalent
HF (MPC) error of -0.071 eV per atom. As in the case of
![]() -point sampling, the CFSE for the MPC interaction is roughly
half that for the Ewald interaction.
-point sampling, the CFSE for the MPC interaction is roughly
half that for the Ewald interaction.
After applying the IPFSE correction obtained from the LDA data the HF
results for
![]() and
and
![]() sampling are very similar. The correspondence of the IPFSE corrected
data for the
sampling are very similar. The correspondence of the IPFSE corrected
data for the ![]() - and
- and ![]() -points demonstrates that to a very
good approximation the IPFSE and CFSE are independent. Estimating the
energy of the infinite system by averaging the energy over a set of
-points demonstrates that to a very
good approximation the IPFSE and CFSE are independent. Estimating the
energy of the infinite system by averaging the energy over a set of
![]() vectors removes the IPFSE but does not remove the CFSE.
vectors removes the IPFSE but does not remove the CFSE.
The CFSE errors from the filled data points in figures 6.1
and 6.2 were fitted to the form ![]() , where
, where ![]() and
and
![]() are parameters and
are parameters and ![]() is the number of electrons in the
simulation cell. The fits give values of
is the number of electrons in the
simulation cell. The fits give values of ![]() in the region of unity
for both the Ewald and MPC interactions. This extrapolation function
is therefore suitable for both interactions, although the size of the
extrapolation term is smaller for the MPC interaction.
in the region of unity
for both the Ewald and MPC interactions. This extrapolation function
is therefore suitable for both interactions, although the size of the
extrapolation term is smaller for the MPC interaction.
![\includegraphics [width=10cm]{Figures/fig1finsize.eps}](pkthimg629.gif)
|
![\includegraphics [width=10cm]{Figures/fig2finsize.eps}](pkthimg630.gif)
|
Single determinant wave functions of Slater-Jastrow form, as
described in chapter 4, were optimised using both the Ewald
and MPC interactions. 32 variable parameters were optimised, and the
resultant wave functions obtained approximately ![]() of the
fixed-node correlation energy. The same pseudopotential as for the LDA
and HF calculations was used.
of the
fixed-node correlation energy. The same pseudopotential as for the LDA
and HF calculations was used.
The wave functions optimised using the different interactions were almost identical. Properties other than the energy, such as pair correlation functions, are therefore hardly affected by the choice of interaction. As the MPC interaction gives the correct interaction between the electrons at short distances it may give a better account of, for example, the short distance behaviour of the pair correlation function, but this point was not investigated.
In figure 6.3 VMC results for energies of the same systems as
in the previous section are shown.6.2 The total energies were calculated to a
statistical accuracy of ![]() eV per atom. The VMC data display
a smaller CFSE than the HF data, probably because the HF exchange hole
is longer ranged than the screened hole obtained in the correlated
calculations. For
eV per atom. The VMC data display
a smaller CFSE than the HF data, probably because the HF exchange hole
is longer ranged than the screened hole obtained in the correlated
calculations. For ![]() = 2, the MPC interaction reduces the VMC finite
size error by more than 50%, from
= 2, the MPC interaction reduces the VMC finite
size error by more than 50%, from ![]() to
to ![]() eV per atom.
eV per atom.
![\includegraphics [width=10cm]{Figures/fig3finsize.eps}](pkthimg634.gif)
|
The MPC interaction is more complicated to apply in DMC than in VMC
because the evaluation of the importance sampled Green's function
requires the local energy. The modified Hamiltonian, and hence the
local energy, depends on the charge density, and therefore the charge
density must be known before the DMC calculation. Fortunately the
``exact'' DMC charge density is not required because the local energy
is relatively insensitive to the charge density used in the
Hamiltonian (equation 6.8) because
![]() is small when
is small when ![]() is small.
is small.
Two candidate charge densities are the charge density of the
determinantal part of the QMC wave function and the charge density of
the VMC guiding wave function. Even for a small system (![]() = 2),
where the potential errors are largest, it was found to be sufficient
to use the LDA charge density during the calculation of the Green's
function and to re-evaluate the charge density dependent term in the
interaction energy using the DMC density obtained at the end of the
calculation. The insensitivity of the Green's function to the charge
density used in the Hamiltonian was tested by running calculations
with LDA, VMC and ``self-consistent'' DMC densities. The sensitivity
rapidly reduces with increasing system size. Using an LDA density
gives errors of less than 0.03 eV per atom for
= 2),
where the potential errors are largest, it was found to be sufficient
to use the LDA charge density during the calculation of the Green's
function and to re-evaluate the charge density dependent term in the
interaction energy using the DMC density obtained at the end of the
calculation. The insensitivity of the Green's function to the charge
density used in the Hamiltonian was tested by running calculations
with LDA, VMC and ``self-consistent'' DMC densities. The sensitivity
rapidly reduces with increasing system size. Using an LDA density
gives errors of less than 0.03 eV per atom for ![]() = 2, and less than
0.01 eV per atom for larger system sizes. Therefore, the requirement
of having a good approximation to the charge density in advance of the
DMC calculation does not pose a significant difficulty. A successful
DMC calculation requires a good quality VMC trial function and its
charge density can be obtained during the process of wave function
optimization.
= 2, and less than
0.01 eV per atom for larger system sizes. Therefore, the requirement
of having a good approximation to the charge density in advance of the
DMC calculation does not pose a significant difficulty. A successful
DMC calculation requires a good quality VMC trial function and its
charge density can be obtained during the process of wave function
optimization.
The DMC calculations were performed using the optimised wavefunctions of
the previous section. A time step of 0.01 a.u. was used. Li et
al. [97] found that using a time step of 0.015
a.u. gave a time-step error of less than 0.03 eV per atom in silicon,
so that our time step error should be even smaller. Total energies were
calculated to a statistical accuracy of ![]() eV per atom.
eV per atom.
In figure 6.4 the results of DMC calculations are shown. The results include a correction for the IPFSE. The convergence behaviour is very similar to the VMC data. The MPC energies are always above those for the Ewald interaction and the MPC interaction significantly reduces the CFSE. These results demonstrate that the finite size errors within VMC and DMC calculations are very similar and that the MPC interaction is similarly effective in both methods, although a little more complex to apply in DMC.
![\includegraphics [width=10cm]{Figures/fig4finsize.eps}](pkthimg638.gif)
|
The residual CFSE errors in both the VMC and DMC calculations are
reasonably fit by a function to the form ![]() . The fit gives a
value of
. The fit gives a
value of ![]() close to unity for both the Ewald and MPC data. For
these data it is possible to obtain more accurate approximations to
close to unity for both the Ewald and MPC data. For
these data it is possible to obtain more accurate approximations to
![]() by using such an extrapolation function,
although the calculations for the large system sizes are costly,
especially within DMC. The extrapolated energies should be more
accurate for the MPC interaction because the extrapolation term is
significantly smaller. The quality of the fit and hence extrapolation
cannot be accurately determined due to the limited number of data
points and statistical errors present in the data.
by using such an extrapolation function,
although the calculations for the large system sizes are costly,
especially within DMC. The extrapolated energies should be more
accurate for the MPC interaction because the extrapolation term is
significantly smaller. The quality of the fit and hence extrapolation
cannot be accurately determined due to the limited number of data
points and statistical errors present in the data.
Many interesting applications of VMC and DMC methods will be to problems in which the quantity of physical interest is the difference in energy between two large systems. Examples of such problems are calculations of excitation energies and defect energies in solids. In such cases the energy of interest is approximately independent of the size of the simulation cell, so that for each simulation cell size it is the energy of the whole simulation cell which must be converged to the required tolerance, not the energy per atom as we plotted in figures 6.3 and 6.4. In these cases extrapolation will be so costly that it can hardly be contemplated. In some cases the CFSE will largely cancel between the two systems, as occurs in excitation energy calculations (see next section). This cancellation cannot always be relied upon, particularly when the simulation cells contain different numbers of particles or different supercell geometries. The use of the MPC interaction should be particularly beneficial in such cases.