The quasiparticle excitation energies are the energies for adding an electron to the system or subtracting one from it. A quasiparticle energy has both real and imaginary parts, the latter giving the quasiparticle lifetime. For the minimum energy electron and hole quasiparticle excitations the imaginary parts of the quasiparticle energies are zero and the quasiparticles have an infinite lifetime. In this case the exact quasiparticle energy gap can be written as
| (6.16) |
where ![]() ,
, ![]() , and
, and ![]() are the ground state
total energies of the
are the ground state
total energies of the ![]() ,
, ![]() and
and ![]() electron
systems. [3] A general
quasiparticle energy gap cannot be written in terms of differences
between energies of exact eigenstates of the system, but such an
approximation is often accurate for low energy gaps. The
quasiparticle energies are measured in photoemission and inverse
photoemission experiments. In an optical absorption experiment a
different process occurs in which an electron is excited from the
valence to the conduction band. This introduces two quasiparticles
into the system, the electron and hole, which interact and can form an
exciton, in which case the lowest excitation energy is smaller than
electron
systems. [3] A general
quasiparticle energy gap cannot be written in terms of differences
between energies of exact eigenstates of the system, but such an
approximation is often accurate for low energy gaps. The
quasiparticle energies are measured in photoemission and inverse
photoemission experiments. In an optical absorption experiment a
different process occurs in which an electron is excited from the
valence to the conduction band. This introduces two quasiparticles
into the system, the electron and hole, which interact and can form an
exciton, in which case the lowest excitation energy is smaller than
![]() by the exciton binding energy,
by the exciton binding energy, ![]() .
.
The HF method gives approximations to the energies of quasiparticles and the interactions between them. According to Koopmans' theorem, if orbital relaxation is neglected, the quasiparticle energies are equal to the HF eigenvalues. The extended Koopmans' theorem [98,99] has been used in conjunction with VMC methods to calculate quasiparticle energies in silicon (see chapter 7 and reference [100]). In both of these methods the ``quasiparticle energies'' are real as they are obtained as approximations to the energy differences between exact eigenstates of the system.
Recently there has been significant progress in applying QMC
techniques to calculate approximate excitation energies from
eigenstates using ``direct methods''. In these approaches an
excitation energy is obtained by performing separate QMC calculations
for the ground and excited states. A Slater-Jastrow wave function is
used for the ground state, and for the optical gap the excited state
is formed by replacing a valence band single particle orbital by a
conduction band one. This calculation is referred to as a
``promotion'' calculation, and such calculations have been reported
for a nitrogen solid [42],
diamond [101,102], and
silicon [103]. Photoemission/inverse
photoemission gaps may be obtained by using QMC to calculate the
ground state energies of the ![]() ,
, ![]() , and
, and ![]() electron
systems. Wave functions for the
electron
systems. Wave functions for the ![]() and
and ![]() electron systems
may be formed by adding or subtracting an orbital from the up- or
down-spin determinants of the
electron systems
may be formed by adding or subtracting an orbital from the up- or
down-spin determinants of the ![]() -electron wave function - an
``addition/subtraction'' calculation. For calculations with periodic
boundary conditions the simulation cell is made charge neutral by
adding a compensating uniform background charge density. Calculations
of this type have been reported for one- [104] and
two-dimensional [92] model systems, while the
results results for a three-dimensional system (silicon) are reported
in this thesis.
-electron wave function - an
``addition/subtraction'' calculation. For calculations with periodic
boundary conditions the simulation cell is made charge neutral by
adding a compensating uniform background charge density. Calculations
of this type have been reported for one- [104] and
two-dimensional [92] model systems, while the
results results for a three-dimensional system (silicon) are reported
in this thesis.
QMC calculations of excitation energies in extended systems are
computationally very demanding because they are `![]() '
effects, i.e., the fractional change in energy is inversely
proportional to the number of electrons in the system. This means
that high statistical accuracy is required to obtain good results.
The largest system for which excitation energies have been calculated
prior to this work is 16 atoms (64
electrons). [103] The total finite size error in
the ground state energy for that system was estimated to be about 16
eV per simulation cell, while the energy scale of interest for the
excitations is of order 0.1 eV. Like almost all methods for
calculating excitation energies, QMC calculations of this type only
work because of a strong cancellation of errors between the ground and
excited states. Finite size errors tend to reduce the energy gap,
while the errors in the trial wave functions are usually larger for
the excited states than for the ground state and so increase the
energy gap. Although good agreement with experimental excitation
energies has been found using small simulation
cells, [101,102,103]
one is left with the suspicion that if larger simulation cells were
used the agreement with experiment might be significantly worse
because the finite size effects would be smaller.
'
effects, i.e., the fractional change in energy is inversely
proportional to the number of electrons in the system. This means
that high statistical accuracy is required to obtain good results.
The largest system for which excitation energies have been calculated
prior to this work is 16 atoms (64
electrons). [103] The total finite size error in
the ground state energy for that system was estimated to be about 16
eV per simulation cell, while the energy scale of interest for the
excitations is of order 0.1 eV. Like almost all methods for
calculating excitation energies, QMC calculations of this type only
work because of a strong cancellation of errors between the ground and
excited states. Finite size errors tend to reduce the energy gap,
while the errors in the trial wave functions are usually larger for
the excited states than for the ground state and so increase the
energy gap. Although good agreement with experimental excitation
energies has been found using small simulation
cells, [101,102,103]
one is left with the suspicion that if larger simulation cells were
used the agreement with experiment might be significantly worse
because the finite size effects would be smaller.
In the next sections, the key questions as to the the size and origin of finite size effects in excitation energy calculations and the differences in finite size effects between promotion and addition/subtraction calculations are addressed. Before these QMC techniques can be relied upon for the calculation of excitation energies it is necessary that finite size effects be properly explored, and answers to these questions found.
If orbital relaxation is neglected, the energy
required to excite an electron from the ![]() (occupied) orbital
into the
(occupied) orbital
into the ![]() (unoccupied) orbital is
(unoccupied) orbital is
where
![]() is the charge density from the
is the charge density from the
![]() orbital. The first term is the eigenvalue difference for the
excitation while the second and third terms are the Hartree and
exchange interactions between the electron and hole. Within this
approximation the electron-hole terms go to zero in the limit of an
infinitely large simulation cell. The fourth and fifth terms on the
right hand side are absent if one uses the Ewald interaction instead
of the MPC interaction, i.e.,
orbital. The first term is the eigenvalue difference for the
excitation while the second and third terms are the Hartree and
exchange interactions between the electron and hole. Within this
approximation the electron-hole terms go to zero in the limit of an
infinitely large simulation cell. The fourth and fifth terms on the
right hand side are absent if one uses the Ewald interaction instead
of the MPC interaction, i.e., ![]() is replaced by
is replaced by ![]() . When the
relaxation of the orbitals is neglected these terms also go to zero
when the size of the simulation cell goes to infinity because
. When the
relaxation of the orbitals is neglected these terms also go to zero
when the size of the simulation cell goes to infinity because ![]() tends to
tends to ![]() over most of the simulation cell.
over most of the simulation cell.
The addition/subtraction gap is given by
where ![]() is the HF ground state energy of the
is the HF ground state energy of the
![]() -electron system,
-electron system,
![]() is the energy of the state
with an electron added to the
is the energy of the state
with an electron added to the ![]() (previously unoccupied)
orbital, along with the uniform background charge, and
(previously unoccupied)
orbital, along with the uniform background charge, and
![]() is the energy of the state where an electron is removed from the
is the energy of the state where an electron is removed from the
![]() orbital, along with the background charge. The standard
Koopmans' theorem has been modified and contains two additional terms,
which also occur in the promotion energy,
orbital, along with the background charge. The standard
Koopmans' theorem has been modified and contains two additional terms,
which also occur in the promotion energy, ![]() . These
additional terms, when evaluated using LDA orbitals, are small even
for a small simulation cell (
. These
additional terms, when evaluated using LDA orbitals, are small even
for a small simulation cell (![]() = 2), being in the range
= 2), being in the range ![]() 0.05
eV, and they decrease rapidly with system size. The use of exact HF
orbitals or orbital relaxation is not expected to greatly affect these
results.
0.05
eV, and they decrease rapidly with system size. The use of exact HF
orbitals or orbital relaxation is not expected to greatly affect these
results.
In figure 6.5 the addition/subtraction energies calculated
using equation 6.19 for the
![]() energy gap and the valence band width, calculated with
both the Ewald and MPC interactions, are shown along with the LDA
values. The results for other low-lying excitation energies show
similar behaviour. The promotion energies are not shown
figure 6.5 because they differ from the
addition/subtraction energies only by the exciton binding energy,
which decreases with increasing system size quite rapidly.
Figure 6.5 shows that the HF results for the Ewald and MPC
interactions are very similar. The band width converges by about
energy gap and the valence band width, calculated with
both the Ewald and MPC interactions, are shown along with the LDA
values. The results for other low-lying excitation energies show
similar behaviour. The promotion energies are not shown
figure 6.5 because they differ from the
addition/subtraction energies only by the exciton binding energy,
which decreases with increasing system size quite rapidly.
Figure 6.5 shows that the HF results for the Ewald and MPC
interactions are very similar. The band width converges by about ![]() = 7, but the band gap is still slowly increasing at
= 7, but the band gap is still slowly increasing at ![]() = 12, and the
Ewald and MPC values are not yet equal, which they must be at
convergence. For the largest system size studied (
= 12, and the
Ewald and MPC values are not yet equal, which they must be at
convergence. For the largest system size studied (![]() = 12) the MPC
gap and valence band width are 7.4 eV and 17.7 eV respectively, which
are a little smaller than the HF values of 8.0 eV and 18.9 eV cited in
Ref. [105]. The major reasons for these
differences are the use of LDA wave functions and LDA-derived
pseudopotentials, although as noted above there is clear evidence that
in our calculations the HF energy gap has still not fully converged at
= 12) the MPC
gap and valence band width are 7.4 eV and 17.7 eV respectively, which
are a little smaller than the HF values of 8.0 eV and 18.9 eV cited in
Ref. [105]. The major reasons for these
differences are the use of LDA wave functions and LDA-derived
pseudopotentials, although as noted above there is clear evidence that
in our calculations the HF energy gap has still not fully converged at
![]() . The LDA excitation energies converge very rapidly with system
size.
. The LDA excitation energies converge very rapidly with system
size.
In a recent study of silicon clusters, Ögüt et al. [106] found large differences between the band gap in the LDA eigenvalues and the band gap calculated by electron addition/subtraction. As shown by Franceschetti et al. [107], these differences are due to the charging of the cluster when an electron is added or subtracted, which does not occur in our calculations because a uniform background is added to preserve charge neutrality. The slow convergence of the HF excitation energies apparent in figure 6.5 therefore arises from the exchange energy. Moreover, because the results with the Ewald and MPC interactions are almost the same, the source of the error is not the interaction with the exchange hole, but the shape of the exchange hole. This is because the excitation energy depends on the change in the exchange hole due to the excitation, which is not strongly localised.
![\includegraphics [width=10cm]{Figures/fig5finsize.eps}](pkthimg673.gif)
|
In a set of calculations on a model two-dimensional system using LDA,
![]() , VMC and DMC techniques, Engel et al. [92]
performed a number of VMC calculations with increasing system size and
found that the addition/subtraction gap tended to increase with system
size. This is the same behaviour as found in the present HF
calculations. Engel et al. went on to give an explanation of
this effect. Their explanation was that in the
, VMC and DMC techniques, Engel et al. [92]
performed a number of VMC calculations with increasing system size and
found that the addition/subtraction gap tended to increase with system
size. This is the same behaviour as found in the present HF
calculations. Engel et al. went on to give an explanation of
this effect. Their explanation was that in the ![]() (
(![]() ) electron
systems there is an additional electrostatic energy due to the
interaction of the extra electron (hole) with the additional electron
(hole) in the other simulation cells. Taking into account the
additional compensating uniform background charge that was added to
keep each cell neutral, this additional energy is negative and
therefore the energies
) electron
systems there is an additional electrostatic energy due to the
interaction of the extra electron (hole) with the additional electron
(hole) in the other simulation cells. Taking into account the
additional compensating uniform background charge that was added to
keep each cell neutral, this additional energy is negative and
therefore the energies ![]() and
and ![]() are lower than they
should be. Engel et al. showed that the observed finite size
effect is much smaller than the Madelung energy for point charges, and
to explain this they argued that the effect would be screened by the
response of the other electrons. This argument implies that the
finite size effects in addition/subtraction calculations are larger
than those in promotion calculations.
are lower than they
should be. Engel et al. showed that the observed finite size
effect is much smaller than the Madelung energy for point charges, and
to explain this they argued that the effect would be screened by the
response of the other electrons. This argument implies that the
finite size effects in addition/subtraction calculations are larger
than those in promotion calculations.
A reanalysis of the situation follows. HF calculations are considered for simplicity, where the interaction energy can be divided into Hartree and exchange contributions. The significant underestimation of the HF band gaps of small systems is not due to the Hartree terms, which by construction are the same as for our LDA calculations and give very small finite size effects in the band gaps. The finite size error in the HF gaps therefore arises from the exchange energy. By comparing band gaps calculated with the Ewald and MPC interactions we can see whether the problem lies with the interaction or with the shape of the exchange hole. Because the band gaps calculated with the Ewald and MPC interactions are very similar, the form of the interaction is not the important consideration. Therefore the source of the problem must be the finite size errors present in the shape of the exchange hole. This argument implies that the finite size effects in addition/subtraction calculations are similar to those in promotion calculations. This view is supported by the HF results that have been presented in this section and also by the VMC results of the next section.
In summary, the HF excitation energies calculated with the Ewald and MPC interactions are very similar. Within HF theory the largest finite size error in excitation energies arises from the shape of the exchange hole, which leads to slow convergence with system size. The finite size errors in promotion and addition/subtraction HF calculations are of similar size.
VMC is computationally cheaper than DMC and so excitation energies may
be computed using VMC over a larger range of system sizes. It is
expected that the finite size effects in DMC will follow those in VMC,
as the VMC calculations retrieve about 90% of the fixed-node
correlation energy. The
![]() excitation energies were computed within VMC in silicon for the system
sizes
excitation energies were computed within VMC in silicon for the system
sizes ![]() = 1, 2, 3, 4 using both promotion and addition/subtraction
methods. The calculations were performed with
= 1, 2, 3, 4 using both promotion and addition/subtraction
methods. The calculations were performed with
![]() and the other computational details were the same as for the
ground state calculations. The Jastrow factors were left unchanged
for the excited state. In tests on the
and the other computational details were the same as for the
ground state calculations. The Jastrow factors were left unchanged
for the excited state. In tests on the ![]() = 2 system, separately
optimizing the Jastrow factors for both ground and excited states did
not significantly change the results. The computational cost of the
= 2 system, separately
optimizing the Jastrow factors for both ground and excited states did
not significantly change the results. The computational cost of the
![]() = 4 calculations is very large; an error bar in the excitation
energies of
= 4 calculations is very large; an error bar in the excitation
energies of ![]() eV requires an error bar of
eV requires an error bar of ![]() eV per
electron. Although the computational effort is large, such a study is
necessary to establish the accuracy of QMC excitation energy
calculations.
eV per
electron. Although the computational effort is large, such a study is
necessary to establish the accuracy of QMC excitation energy
calculations.
In figure 6.6 excitation energies obtained with the Ewald
interaction via the promotion and addition/subtraction methods are
shown. Results for the MPC interaction were found to be very similar.
The promotion and addition/subtraction results are nearly the same,
but the promotion energies are slightly smaller because they include
an exciton binding energy, which decreases as the system size
increases. The results are consistent with a slow increase in the
excitation energy with system size and indicate that reasonable
convergence is already obtained at ![]() = 2. The increase in
excitation energies with system size is the same trend as in HF
calculations, although the finite size errors are smaller in the
correlated calculations. The finite size errors in the promotion and
addition/subtraction methods are not significantly different at this
level of statistical accuracy. On general grounds one would expect the
finite size effect in the promotion calculation to be slightly larger.
This follows because the trend for both promotion and
addition/subtraction calculations is that the excitation energy is
reduced for small system sizes and this effect is enhanced in the
promotion calculation by the exciton binding energy which is larger
for small systems. The calculated excitation energy is roughly 4 eV,
which is larger than the experimental value of 3.40
eV, [108] and also a little larger than the
DMC value for an
= 2. The increase in
excitation energies with system size is the same trend as in HF
calculations, although the finite size errors are smaller in the
correlated calculations. The finite size errors in the promotion and
addition/subtraction methods are not significantly different at this
level of statistical accuracy. On general grounds one would expect the
finite size effect in the promotion calculation to be slightly larger.
This follows because the trend for both promotion and
addition/subtraction calculations is that the excitation energy is
reduced for small system sizes and this effect is enhanced in the
promotion calculation by the exciton binding energy which is larger
for small systems. The calculated excitation energy is roughly 4 eV,
which is larger than the experimental value of 3.40
eV, [108] and also a little larger than the
DMC value for an ![]() = 2 simulation cell of 3.7
eV. [103] This study demonstrates that the
largest contribution to the error in the VMC band gap for
= 2 simulation cell of 3.7
eV. [103] This study demonstrates that the
largest contribution to the error in the VMC band gap for ![]() arises from the approximate nature of the trial wave functions, and
not from finite size effects.
arises from the approximate nature of the trial wave functions, and
not from finite size effects.
![\includegraphics [width=10cm]{Figures/fig6finsize.eps}](pkthimg678.gif)
|
The exciton binding energy can be calculated as the difference between
the promotion and addition/subtraction gaps. The exciton binding
energy for the
![]() excitation
is small and it is only statistically resolvable for the
smallest (
excitation
is small and it is only statistically resolvable for the
smallest (![]() ) cell, which gives a value of
) cell, which gives a value of ![]() eV. In
earlier QMC
calculations [42,101,102]
the exciton binding energy was estimated using the Mott-Wannier
formula
eV. In
earlier QMC
calculations [42,101,102]
the exciton binding energy was estimated using the Mott-Wannier
formula
| (6.20) |
where ![]() is the relative permittivity and
is the relative permittivity and ![]() is the
radius of the localisation region. Using
is the
radius of the localisation region. Using ![]() = 11.7 and the
appropriate radius for
= 11.7 and the
appropriate radius for ![]() = 1 of
= 1 of ![]() = 4.0 a.u. gives an exciton
binding energy of 0.29 eV. This is extremely close to the VMC value,
but the excellent agreement is probably fortuitous since the
= 4.0 a.u. gives an exciton
binding energy of 0.29 eV. This is extremely close to the VMC value,
but the excellent agreement is probably fortuitous since the ![]() = 1
cell is so small that it is appropriate to use a value of
= 1
cell is so small that it is appropriate to use a value of ![]() at finite wave vector, which would be smaller. The exciton binding
energy may also be evaluated within HF theory as the sum of the second
and third terms on the r.h.s. of equation 6.18. This gives
0.75 eV for the
at finite wave vector, which would be smaller. The exciton binding
energy may also be evaluated within HF theory as the sum of the second
and third terms on the r.h.s. of equation 6.18. This gives
0.75 eV for the ![]() cell using the Ewald interaction, which is
considerably larger than the VMC value because the latter calculation
includes screening of the electron-electron interaction.
cell using the Ewald interaction, which is
considerably larger than the VMC value because the latter calculation
includes screening of the electron-electron interaction.
Note that the promotion and addition/subtraction methods differ
significantly in the required computational effort. If the intrinsic
variance of the local energy is the same for each of the energies,
which is a good approximation for silicon wavefunctions, then the most
efficient way to achieve the same error bar in an addition/subtraction
gap is to perform ![]() moves for each of the
moves for each of the ![]() and
and ![]() systems
and
systems
and ![]() for the ground state, giving a total cost of
for the ground state, giving a total cost of ![]() moves,
where an acceptable error bar is obtained in a promotion calculation
by performing
moves,
where an acceptable error bar is obtained in a promotion calculation
by performing ![]() Monte Carlo moves for both the ground and excited
states. It is therefore four times more expensive to calculate an
energy gap to some given accuracy by the addition/subtraction method
than by the promotion method.
Monte Carlo moves for both the ground and excited
states. It is therefore four times more expensive to calculate an
energy gap to some given accuracy by the addition/subtraction method
than by the promotion method.
In this section a modified electron-electron interaction specifically
designed to describe excitation energies within periodic boundary
conditions simulations in introduced. Two problems arise when trying
to model excitations using finite simulation cells subject to periodic
boundary conditions. One is that the excitation is ``squeezed'' into
the simulation cell, and the other is that there are spurious
interaction between the periodic replicas of the simulation cell.
Here the problem of the spurious interactions is addressed using the
ideas of the MPC interaction. The charge density on promotion
or addition/subtraction of an electron can be written as the sum of
the ground state charge density and a small deviation, i.e.,
![]() . The
Hartree term can be modified so that this charge density interacts with the
ground state density in the replicas. This leads to the interaction
energy
. The
Hartree term can be modified so that this charge density interacts with the
ground state density in the replicas. This leads to the interaction
energy
where
![]() generates the charge density
generates the charge density
![]() and the ground state charge density,
and the ground state charge density, ![]() , is fixed.
A HF analysis of this interaction shows that the HF equations are
identical to equation 6.17, so that the orbitals and
eigenvalues are unaltered. However, the ground and excited state
energy expressions are modified. For the excited states analogues of
equations 6.18 and 6.19 are obtained, but without
the terms involving
, is fixed.
A HF analysis of this interaction shows that the HF equations are
identical to equation 6.17, so that the orbitals and
eigenvalues are unaltered. However, the ground and excited state
energy expressions are modified. For the excited states analogues of
equations 6.18 and 6.19 are obtained, but without
the terms involving ![]() , i.e., the standard
Koopmans' theorem is retrieved. Although these terms are small
for silicon, they will be significant in cases when the
change in the charge density of the exciton is strongly
localized. This analysis also provides further evidence that the
electrostatic interactions between the simulation cell and its
replicas is not necessarily an important source of finite size error
in excited state energy calculations; for these silicon systems, the
finite size errors in the excitations are small.
, i.e., the standard
Koopmans' theorem is retrieved. Although these terms are small
for silicon, they will be significant in cases when the
change in the charge density of the exciton is strongly
localized. This analysis also provides further evidence that the
electrostatic interactions between the simulation cell and its
replicas is not necessarily an important source of finite size error
in excited state energy calculations; for these silicon systems, the
finite size errors in the excitations are small.
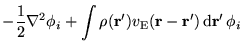
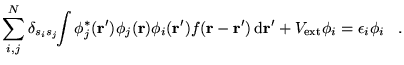
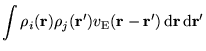
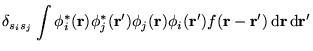
![$\displaystyle \frac{1}{2} \int \rho_i({\bf r}) \rho_i({\bf r}')
\left[ v_{\rm E...
...bf r}-{\bf r}')-f({\bf r}-{\bf r}')\right] \, {\rm d}{\bf r} \, {\rm d}{\bf r}'$](pkthimg658.gif)
![$\displaystyle \frac{1}{2} \int
\rho_j({\bf r}) \rho_j({\bf r}') \left[ v_{\rm E...
...r}-{\bf r}')-f({\bf r}-{\bf r}')\right] \, {\rm d}{\bf r} \, {\rm d}{\bf r}'\;,$](pkthimg659.gif)
![$\displaystyle \frac{1}{2} \int \rho_j({\bf r}) \rho_j({\bf r}') \left[ v_{\rm E...
...{\bf r}')-f({\bf r}-{\bf r}')\right] \, {\rm d}{\bf r} \, {\rm d}{\bf r}' \;\;,$](pkthimg665.gif)
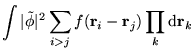
![$\displaystyle \int
\tilde{\rho}({\bf r})\rho({\bf r}') \left[ v_{\rm E}({\bf r}-{\bf r}')- f({\bf r}-{\bf r}') \right] \, {\rm d}{\bf r}\, {\rm d}{\bf r}'$](pkthimg689.gif)
![$\displaystyle \frac{1}{2}\int \rho({\bf r})\rho({\bf r}') \left[
v_{\rm E}({\bf...
...{\bf r}')-f({\bf r}-{\bf r}') \right] \, {\rm d}{\bf r}\, {\rm d}{\bf r}' \;\;,$](pkthimg690.gif)